Beyens Olivier, De Winter Hans
Laboratory of Medicinal Chemistry, Department of Pharmaceutical Sciences, University of Antwerp, Universiteitsplein 1, 2610, Wilrijk, Belgium.
J Cheminform. 2024 Feb 27;16(1):23. doi: 10.1186/s13321-024-00819-y.
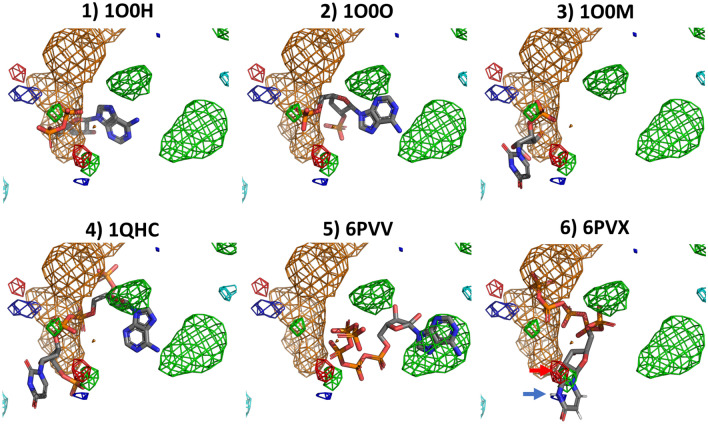
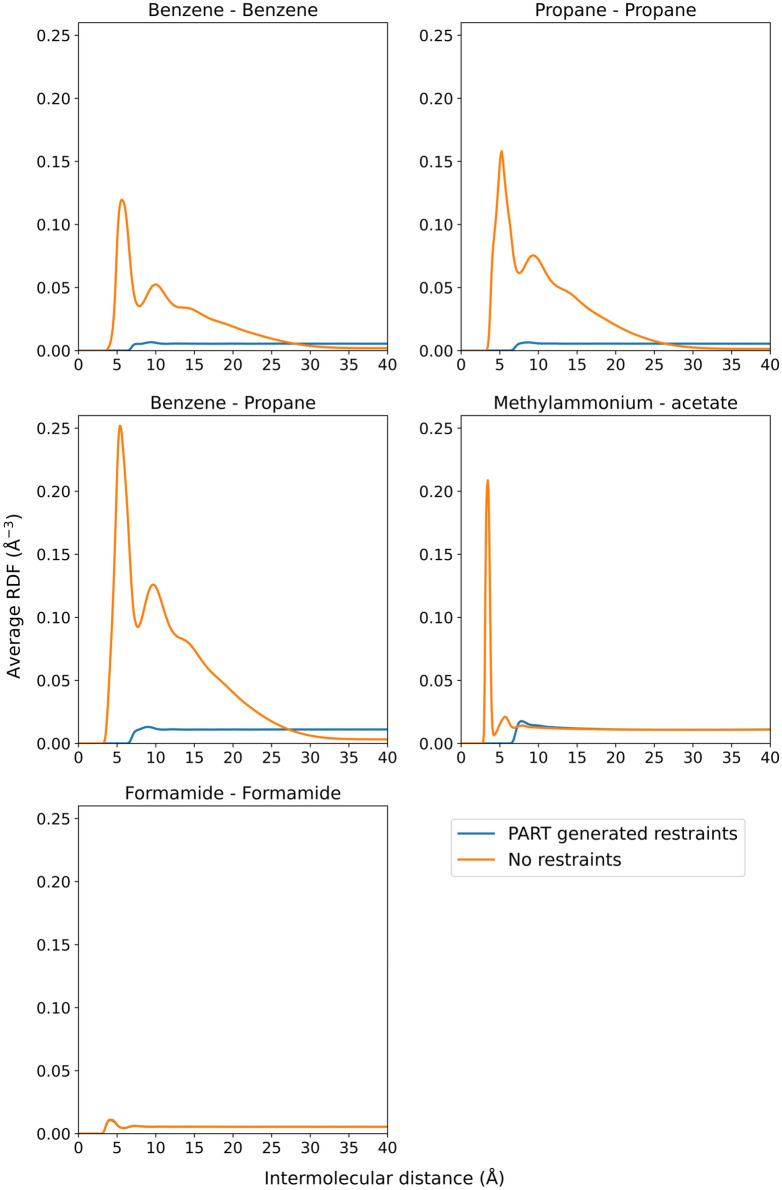
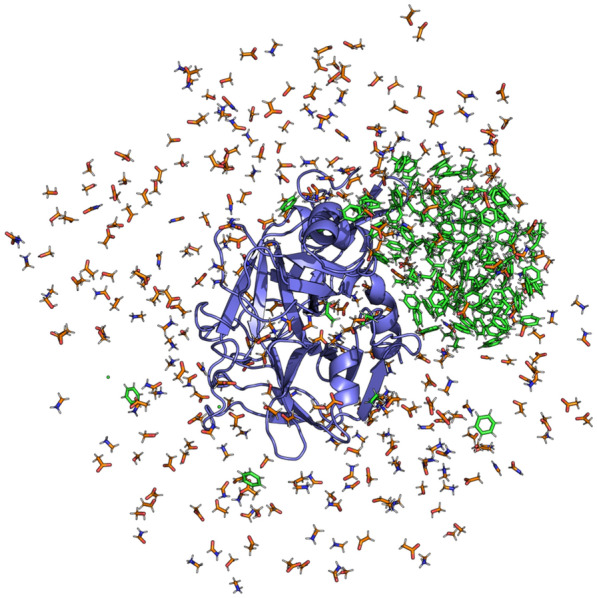
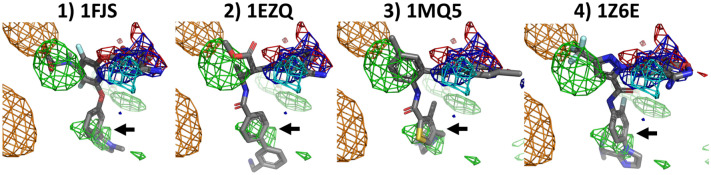
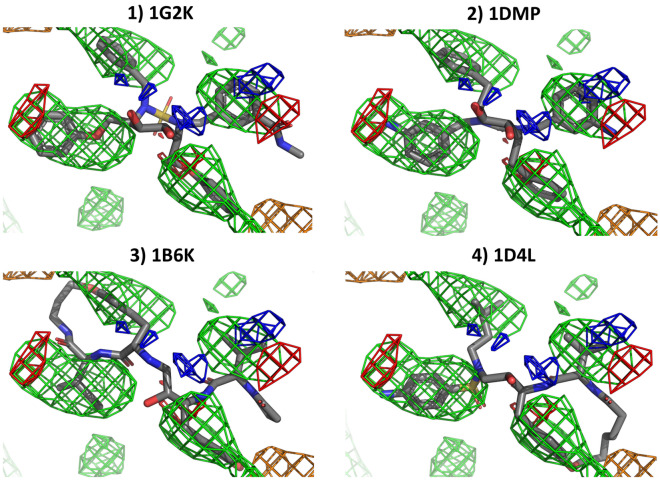
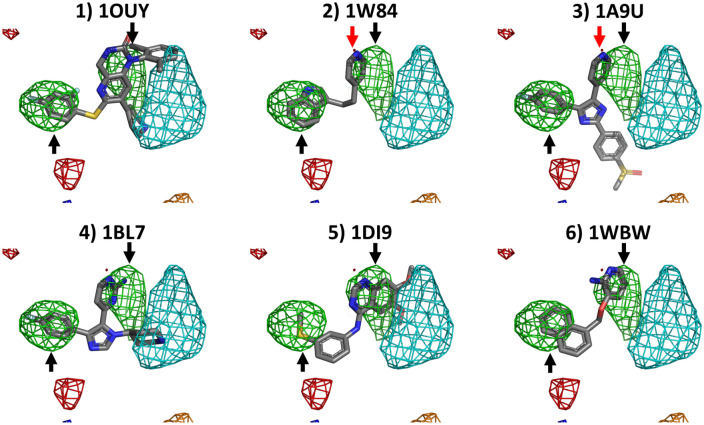
Cosolvent molecular dynamics (MD) simulations are molecular dynamics simulations used to identify preferable locations of small organic fragments on a protein target. Most cosolvent molecular dynamics workflows make use of only water-soluble fragments, as hydrophobic fragments would cause lipophilic aggregation. To date the two approaches that allow usage of hydrophobic cosolvent molecules are to use a low (0.2 M) concentration of hydrophobic probes, with the disadvantage of a lower sampling speed, or to use force field modifications, with the disadvantage of a difficult and inflexible setup procedure. Here we present a third alternative, that does not suffer from low sampling speed nor from cumbersome preparation procedures. We have built an easy-to-use open source command line tool PART (Plumed Automatic Restraining Tool) to generate a PLUMED file handling all intermolecular restraints to prevent lipophilic aggregation. We have compared restrained and unrestrained cosolvent MD simulations, showing that restraints are necessary to prevent lipophilic aggregation at hydrophobic probe concentrations of 0.5 M. Furthermore, we benchmarked PART generated restraints on a test set of four proteins (Factor-Xa, HIV protease, P38 MAP kinase and RNase A), showing that cosolvent MD with PART generated restraints qualitatively reproduces binding features of cocrystallised ligands.
共溶剂分子动力学(MD)模拟是用于确定小有机片段在蛋白质靶点上优选位置的分子动力学模拟。大多数共溶剂分子动力学工作流程仅使用水溶性片段,因为疏水片段会导致亲脂性聚集。迄今为止,允许使用疏水共溶剂分子的两种方法是使用低浓度(0.2 M)的疏水探针,但缺点是采样速度较低;或者使用力场修正,但缺点是设置过程困难且不灵活。在此,我们提出第三种方法,它既不存在采样速度低的问题,也不存在繁琐的制备程序问题。我们构建了一个易于使用的开源命令行工具PART(Plumed自动约束工具)来生成一个PLUMED文件,处理所有分子间约束以防止亲脂性聚集。我们比较了受约束和不受约束的共溶剂MD模拟,结果表明在0.5 M的疏水探针浓度下,约束对于防止亲脂性聚集是必要的。此外,我们在一组包含四种蛋白质(凝血因子Xa、HIV蛋白酶、P38丝裂原活化蛋白激酶和核糖核酸酶A)的测试集上对PART生成的约束进行了基准测试,结果表明使用PART生成约束的共溶剂MD能够定性地重现共结晶配体的结合特征。