Department of Gastroenterology and Hepatology and Sichuan University-University of Oxford Huaxi Joint Centre for Gastrointestinal Cancer, West China Hospital, Sichuan University, #37 Guoxue Road, Chengdu, 610041, Sichuan, China.
J Transl Med. 2024 Feb 29;22(1):214. doi: 10.1186/s12967-024-05029-6.
Primary biliary cholangitis (PBC) and autoimmune hepatitis (AIH) variant syndrome (VS) exhibit a complex overlap of AIH features with PBC, leading to poorer prognoses than those with PBC or AIH alone. The biomarkers associated with drug response and potential molecular mechanisms in this syndrome have not been fully elucidated.
Whole-transcriptome sequencing was employed to discern differentially expressed (DE) RNAs within good responders (GR) and poor responders (PR) among patients with PBC/AIH VS. Subsequent gene ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis were conducted for the identified DE RNAs. Plasma metabolomics was employed to delineate the metabolic profiles distinguishing PR and GR groups. The quantification of immune cell profiles and associated cytokines was achieved through flow cytometry and immunoassay technology. Uni- and multivariable logistic regression analyses were conducted to construct a predictive model for insufficient biochemical response. The performance of the model was assessed by computing the area under the receiver operating characteristic (AUC) curve, sensitivity, and specificity.
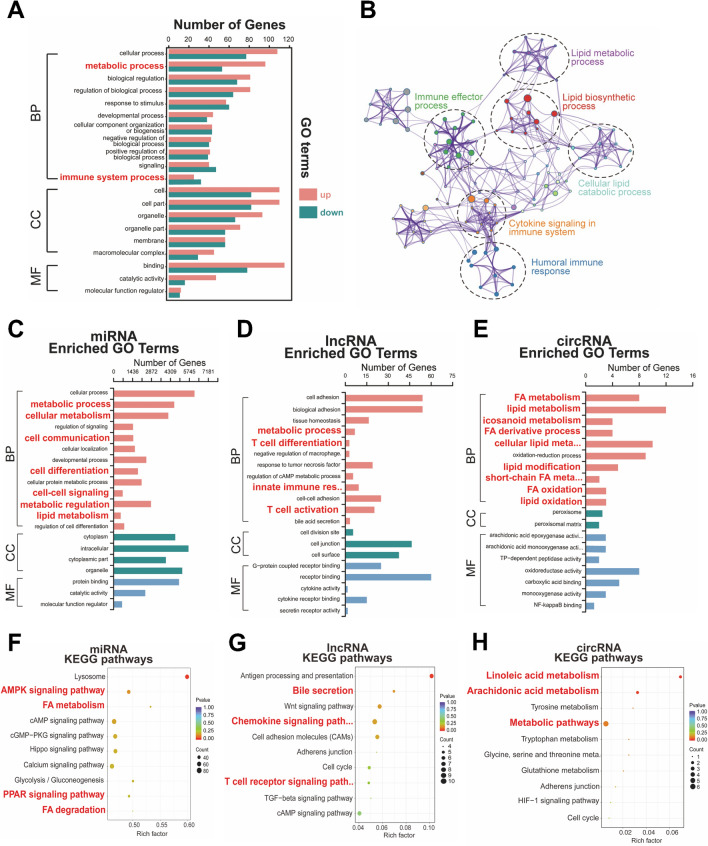
The analysis identified 224 differentially expressed (DE) mRNAs, 189 DE long non-coding RNAs, 39 DE circular RNAs, and 63 DE microRNAs. Functional pathway analysis revealed enrichment in lipid metabolic pathways and immune response. Metabolomics disclosed dysregulated lipid metabolism and identified PC (18:2/18:2) and PC (16:0/20:3) as predictors. CD4 T helper (Th) cells, including Th2 cells and regulatory T cells (Tregs), were upregulated in the GR group. Pro-inflammatory cytokines (IFN-γ, TNF-α, IL-9, and IL-17) were downregulated in the GR group, while anti-inflammatory cytokines (IL-10, IL-4, IL-5, and IL-22) were elevated. Regulatory networks were constructed, identifying CACNA1H and ACAA1 as target genes. A predictive model based on these indicators demonstrated an AUC of 0.986 in the primary cohort and an AUC of 0.940 in the validation cohort for predicting complete biochemical response.
A combined model integrating genomic, metabolic, and cytokinomic features demonstrated high accuracy in predicting insufficient biochemical response in patients with PBC/AIH VS. Early recognition of individuals at elevated risk for insufficient response allows for the prompt initiation of additional treatments.
原发性胆汁性胆管炎(PBC)和自身免疫性肝炎(AIH)变异综合征(VS)表现为 AIH 特征与 PBC 的复杂重叠,导致预后比单纯 PBC 或 AIH 更差。与该综合征中药物反应相关的生物标志物和潜在分子机制尚未完全阐明。
采用全转录组测序技术,识别 PBC/AIH VS 患者中良好反应者(GR)和不良反应者(PR)之间差异表达的(DE)RNA。对鉴定出的 DE RNA 进行基因本体(GO)分析和京都基因与基因组百科全书(KEGG)通路分析。采用血浆代谢组学描绘区分 PR 和 GR 组的代谢谱。通过流式细胞术和免疫测定技术定量检测免疫细胞谱和相关细胞因子。采用单变量和多变量逻辑回归分析构建预测生化反应不足的模型。通过计算接收者操作特征(ROC)曲线下的面积(AUC)、敏感性和特异性来评估模型的性能。
分析确定了 224 个差异表达(DE)mRNA、189 个 DE 长链非编码 RNA、39 个 DE 环状 RNA 和 63 个 DE microRNA。功能途径分析显示脂质代谢途径和免疫反应富集。代谢组学揭示了脂质代谢失调,并鉴定出 PC(18:2/18:2)和 PC(16:0/20:3)作为预测因子。GR 组中 CD4 辅助性 T 细胞(包括 Th2 细胞和调节性 T 细胞[Tregs])上调。GR 组中促炎细胞因子(IFN-γ、TNF-α、IL-9 和 IL-17)下调,而抗炎细胞因子(IL-10、IL-4、IL-5 和 IL-22)上调。构建了调控网络,鉴定出 CACNA1H 和 ACAA1 作为靶基因。基于这些指标的预测模型在初步队列中的 AUC 为 0.986,在验证队列中的 AUC 为 0.940,用于预测完全生化反应。
整合基因组、代谢组和细胞因子组学特征的综合模型在预测 PBC/AIH VS 患者生化反应不足方面具有很高的准确性。早期识别对生化反应不足风险较高的个体,可及时开始额外的治疗。