Kandel Sangam, Hartzell Susanna L, Ingold Ashton K, Turner Grace A, Kennedy Joshua L, Ussery David W
Department of Biomedical Informatics, University of Arkansas for Medical Sciences, Little Rock, AR, United States.
Arkansas Children's Research Institute, Little Rock, AR, United States.
Front Microbiol. 2024 Feb 14;15:1272972. doi: 10.3389/fmicb.2024.1272972. eCollection 2024.
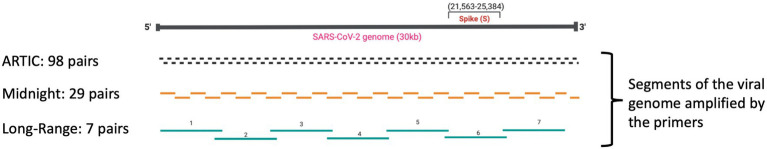
Whole Genome Sequencing (WGS) of the SARS-CoV-2 virus is crucial in the surveillance of the COVID-19 pandemic. Several primer schemes have been developed to sequence nearly all of the ~30,000 nucleotide SARS-CoV-2 genome, using a multiplex PCR approach to amplify cDNA copies of the viral genomic RNA. Midnight primers and ARTIC V4.1 primers are the most popular primer schemes that can amplify segments of SARS-CoV-2 (400 bp and 1200 bp, respectively) tiled across the viral RNA genome. Mutations within primer binding sites and primer-primer interactions can result in amplicon dropouts and coverage bias, yielding low-quality genomes with 'Ns' inserted in the missing amplicon regions, causing inaccurate lineage assignments, and making it challenging to monitor lineage-specific mutations in Variants of Concern (VoCs).
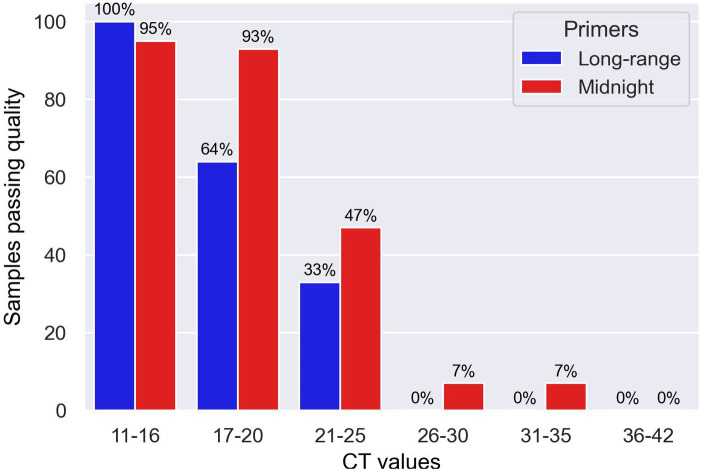
In this study we used a set of seven long-range PCR primer pairs to sequence clinical isolates of SARS-CoV-2 on Oxford Nanopore sequencer. These long-range primers generate seven amplicons approximately 4500 bp that covered whole genome of SARS-CoV-2. One of these regions includes the full-length S-gene by using a set of flanking primers. We also evaluated the performance of these long-range primers with Midnight primers by sequencing 94 clinical isolates in a Nanopore flow cell.
Using a small set of long-range primers to sequence SARS-CoV-2 genomes reduces the possibility of amplicon dropout and coverage bias. The key finding of this study is that long range primers can be used in single-molecule sequencing of RNA viruses in surveillance of emerging variants. We also show that by designing primers flanking the S-gene, we can obtain reliable identification of SARS-CoV-2 variants.
严重急性呼吸综合征冠状病毒2(SARS-CoV-2)病毒的全基因组测序对于新冠疫情的监测至关重要。已经开发了几种引物方案,通过多重聚合酶链反应(PCR)方法扩增病毒基因组RNA的互补DNA(cDNA)拷贝,来对近30,000个核苷酸的SARS-CoV-2基因组几乎全部进行测序。午夜引物和ARTIC V4.1引物是最常用的引物方案,它们可以扩增沿着病毒RNA基因组排列的SARS-CoV-2片段(分别为400碱基对和1200碱基对)。引物结合位点内的突变以及引物-引物相互作用可能导致扩增子缺失和覆盖偏差,产生在缺失的扩增子区域插入“N”的低质量基因组,导致谱系分配不准确,并使得监测关注变异株(VoC)中的谱系特异性突变具有挑战性。
在本研究中,我们使用了一组七个长程PCR引物对,在牛津纳米孔测序仪上对SARS-CoV-2临床分离株进行测序。这些长程引物产生大约4500碱基对的七个扩增子,覆盖了SARS-CoV-2的全基因组。其中一个区域通过使用一组侧翼引物包含全长S基因。我们还通过在纳米孔流动池中对94个临床分离株进行测序,用午夜引物评估了这些长程引物的性能。
使用一小组长程引物对SARS-CoV-2基因组进行测序可降低扩增子缺失和覆盖偏差的可能性。本研究的关键发现是,长程引物可用于新兴变异株监测中RNA病毒的单分子测序。我们还表明,通过设计S基因侧翼的引物,我们可以可靠地鉴定SARS-CoV-2变异株。