Passchier Emma M J, Bisseling Quinty, Helman Guy, van Spaendonk Rosalina M L, Simons Cas, Olsthoorn René C L, van der Veen Hieke, Abbink Truus E M, van der Knaap Marjo S, Min Rogier
Department of Child Neurology, Amsterdam Leukodystrophy Center, Emma Children's Hospital, Amsterdam University Medical Center, Amsterdam Neuroscience, Amsterdam, Netherlands.
Department of Integrative Neurophysiology, Center for Neurogenomics and Cognitive Research, Vrije Universiteit Amsterdam, Amsterdam Neuroscience, Amsterdam, Netherlands.
Front Genet. 2024 Feb 29;15:1352947. doi: 10.3389/fgene.2024.1352947. eCollection 2024.
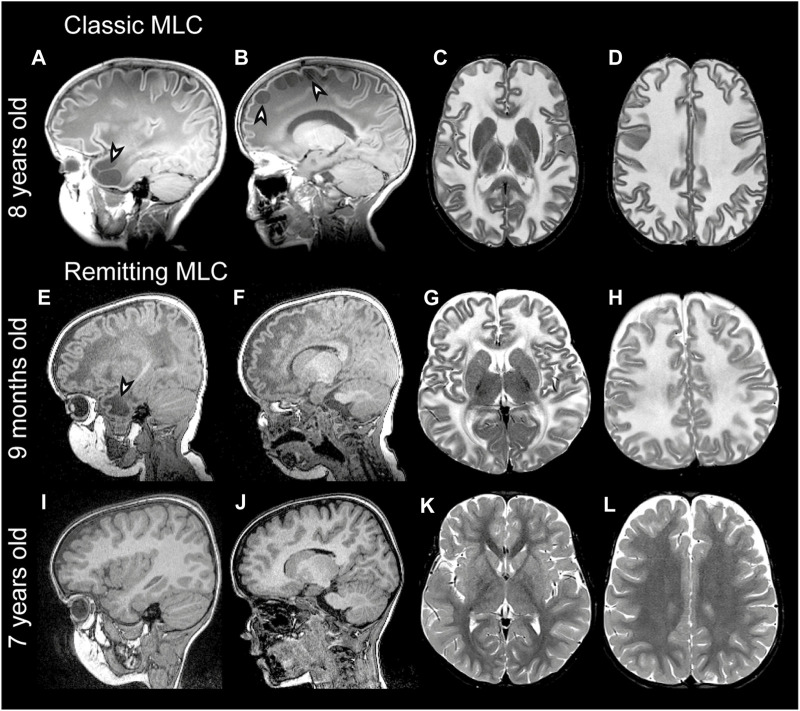
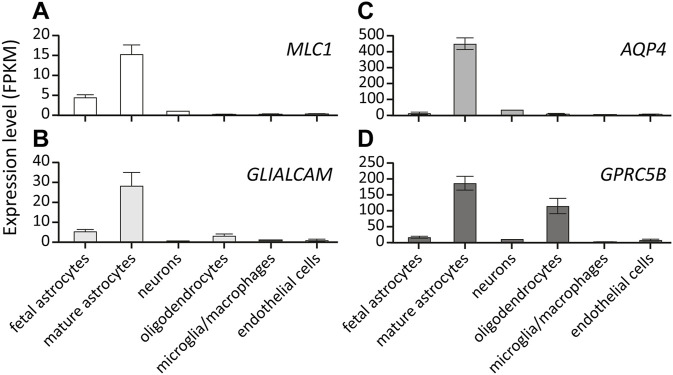
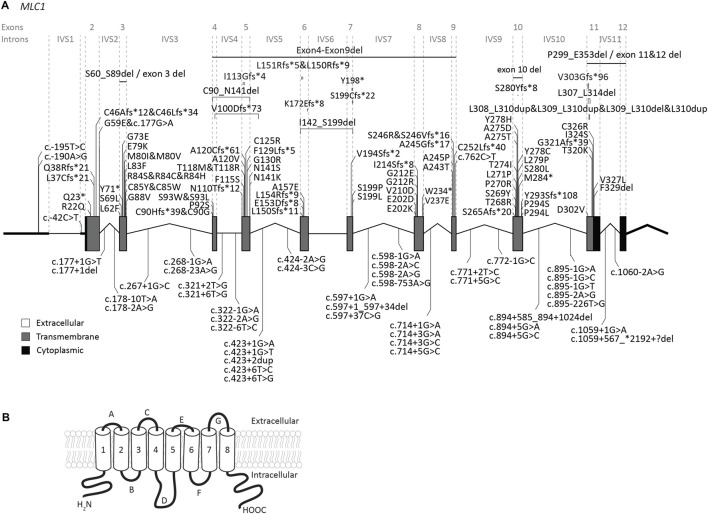
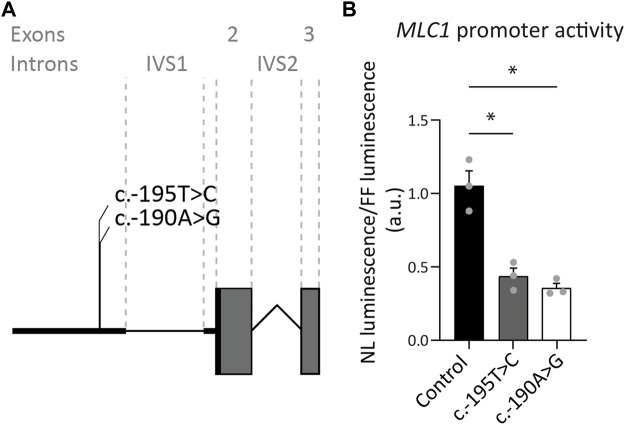
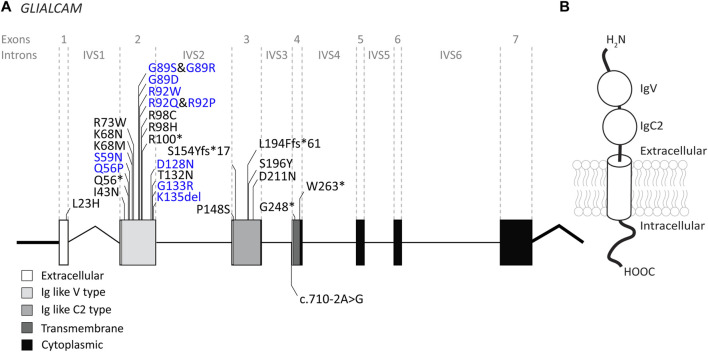
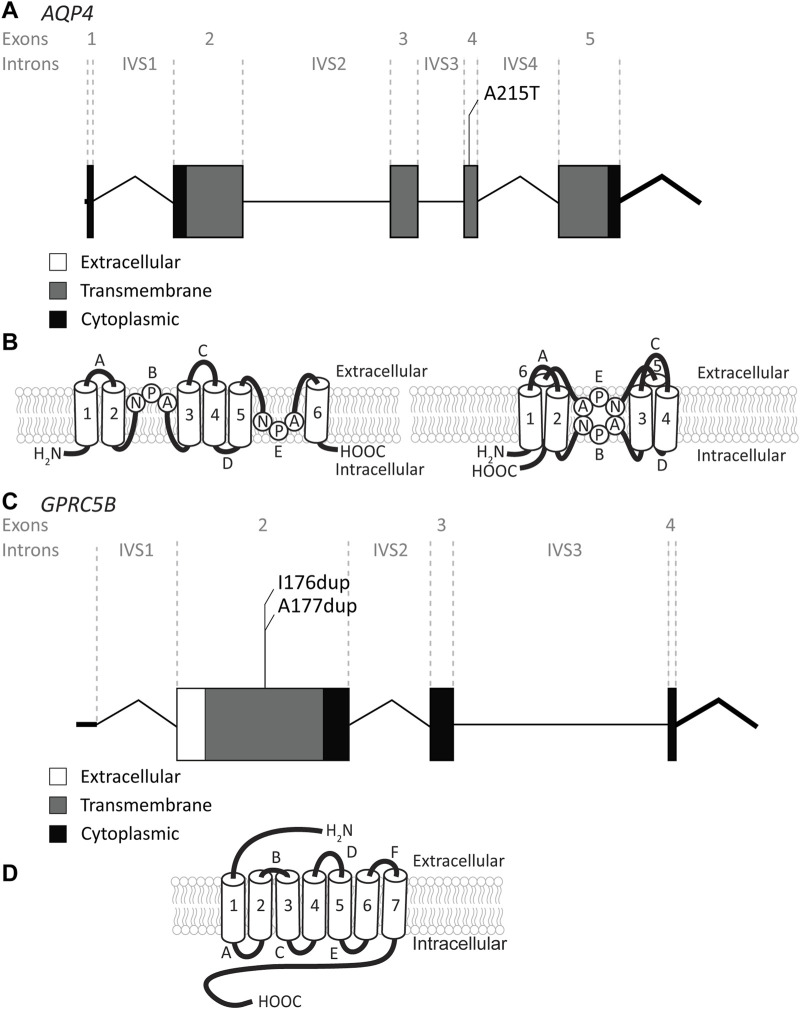
The leukodystrophy megalencephalic leukoencephalopathy with subcortical cysts (MLC) is characterized by infantile-onset macrocephaly and chronic edema of the brain white matter. With delayed onset, patients typically experience motor problems, epilepsy and slow cognitive decline. No treatment is available. Classic MLC is caused by bi-allelic recessive pathogenic variants in or (also called ). Heterozygous dominant pathogenic variants in lead to remitting MLC, where patients show a similar phenotype in early life, followed by normalization of white matter edema and no clinical regression. Rare patients with heterozygous dominant variants in and classic MLC were recently described. In addition, two siblings with bi-allelic recessive variants in and remitting MLC have been identified. The last systematic overview of variants linked to MLC dates back to 2006. We provide an updated overview of published and novel variants. We report on genetic variants from 508 patients with MLC as confirmed by MRI diagnosis (258 from our database and 250 extracted from 64 published reports). We describe 151 unique variants, 29 variants, 2 variants and 1 variant observed in these MLC patients. We include experiments confirming pathogenicity for some variants, discuss particularly notable variants, and provide an overview of recent scientific and clinical insight in the pathophysiology of MLC.
巨脑性白质脑病伴皮质下囊肿(MLC)这一脑白质营养不良症的特征为婴儿期发病的巨头畸形和脑白质慢性水肿。起病较晚时,患者通常会出现运动问题、癫痫和认知功能缓慢衰退。目前尚无有效治疗方法。经典型MLC由MAG或MAGT1(也称为MAGUK)双等位基因隐性致病变异引起。MAG杂合显性致病变异会导致缓解型MLC,患者在生命早期表现出相似的表型,随后脑白质水肿恢复正常且无临床症状恶化。最近报道了罕见的具有MAG杂合显性变异且临床表现类似经典型MLC的患者。此外,还鉴定出了两名具有MAGT1双等位基因隐性变异且临床表现为缓解型MLC的同胞。上一次与MLC相关变异的系统综述可追溯到2006年。我们提供了已发表和新发现变异的最新综述。我们报告了508例经MRI诊断确诊的MLC患者的基因变异情况(258例来自我们的数据库,250例从64篇已发表报告中提取)。我们描述了在这些MLC患者中观察到的151个独特的MAG变异、29个MAGT1变异、2个GPR179变异和1个TMEM106B变异。我们纳入了一些确认某些变异致病性的实验,讨论了特别值得注意的变异,并概述了近期关于MLC病理生理学的科学和临床见解。