Cardio-CARE, Medizincampus Davos, Herman-Burchard-Str. 1, Davos Wolfgang, 7265, Davos, Switzerland.
Swiss Institute of Bioinformatics, Lausanne, Switzerland.
Hum Genet. 2024 May;143(5):625-634. doi: 10.1007/s00439-024-02670-5. Epub 2024 Apr 4.
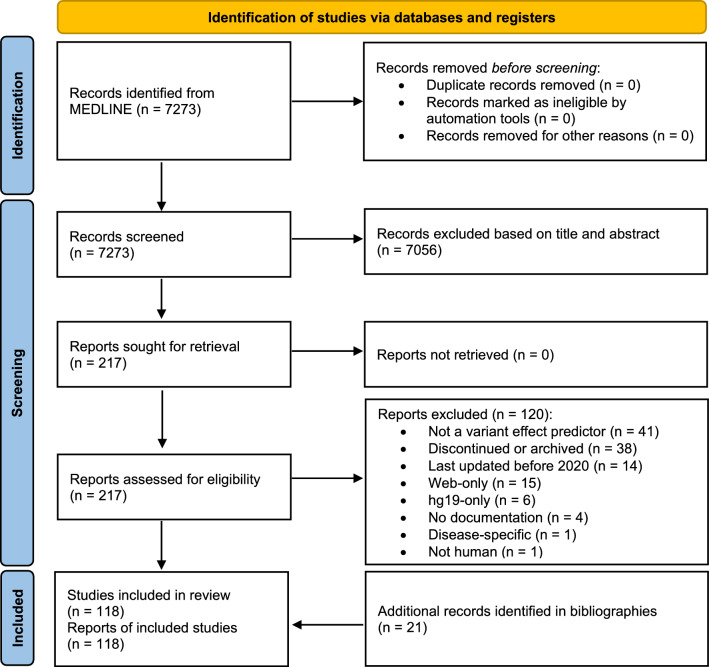
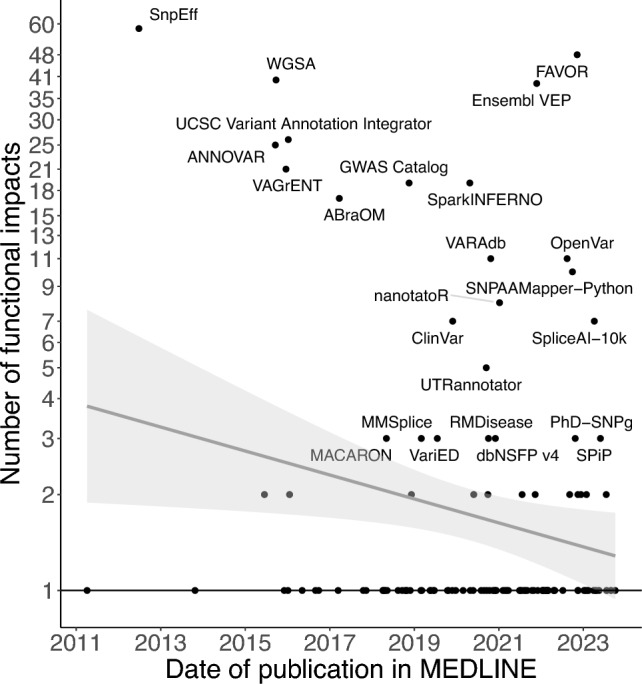
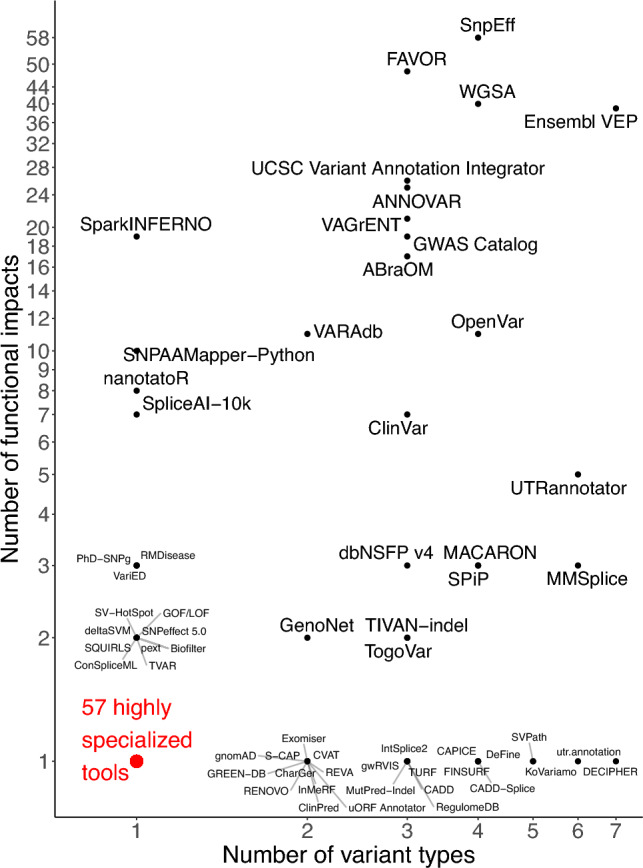
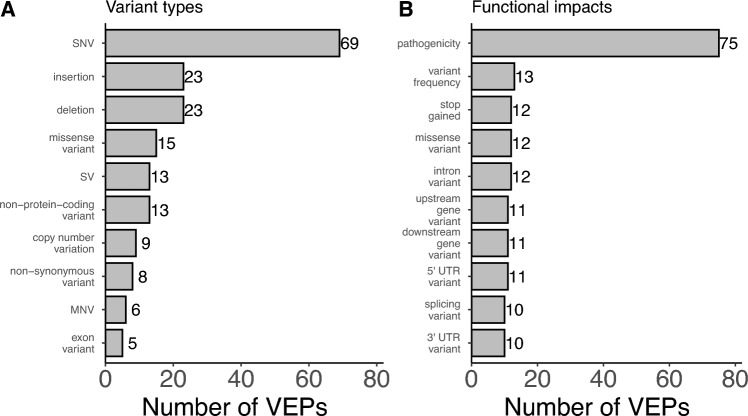
Large-scale association analyses using whole-genome sequence data have become feasible, but understanding the functional impacts of these associations remains challenging. Although many tools are available to predict the functional impacts of genetic variants, it is unclear which tool should be used in practice. This work provides a practical guide to assist in selecting appropriate tools for variant annotation. We conducted a MEDLINE search up to November 10, 2023, and included tools that are applicable to a broad range of phenotypes, can be used locally, and have been recently updated. Tools were categorized based on the types of variants they accept and the functional impacts they predict. Sequence Ontology terms were used for standardization. We identified 118 databases and software packages, encompassing 36 variant types and 161 functional impacts. Combining only three tools, namely SnpEff, FAVOR, and SparkINFERNO, allows predicting 99 (61%) distinct functional impacts. Thirty-seven tools predict 89 functional impacts that are not supported by any other tool, while 75 tools predict pathogenicity and can be used within the ACMG/AMP guidelines in a clinical context. We launched a website allowing researchers to select tools based on desired variants and impacts. In summary, more than 100 tools are already available to predict approximately 160 functional impacts. About 60% of the functional impacts can be predicted by the combination of three tools. Unexpectedly, recent tools do not predict more impacts than older ones. Future research should allow predicting the functionality of so far unsupported variant types, such as gene fusions.URL: https://cardio-care.shinyapps.io/VEP_Finder/ .Registration: OSF Registries on November 10, 2023, https://osf.io/s2gct .
大规模的全基因组关联分析已经成为可能,但理解这些关联的功能影响仍然具有挑战性。虽然有许多工具可用于预测遗传变异的功能影响,但在实践中不清楚应该使用哪种工具。本研究提供了一个实用指南,以协助选择适当的工具进行变异注释。我们进行了截至 2023 年 11 月 10 日的 MEDLINE 检索,并纳入了适用于广泛表型、可本地使用且最近更新的工具。工具根据它们接受的变异类型和预测的功能影响进行分类。使用序列本体论术语进行标准化。我们确定了 118 个数据库和软件包,涵盖 36 种变异类型和 161 种功能影响。仅结合使用三种工具,即 SnpEff、FAVOR 和 SparkINFERNO,就可以预测 99 种(61%)不同的功能影响。37 种工具预测了 75 种工具不支持的 89 种功能影响,而 75 种工具预测了致病性,并且可以在临床背景下使用 ACMG/AMP 指南。我们推出了一个网站,允许研究人员根据所需的变异和影响选择工具。总之,已经有 100 多种工具可用于预测大约 160 种功能影响。约 60%的功能影响可以通过三种工具的组合来预测。出乎意料的是,最近的工具并没有比旧的工具预测更多的影响。未来的研究应该允许预测迄今为止不支持的变异类型的功能,例如基因融合。网址:https://cardio-care.shinyapps.io/VEP_Finder/ 。注册:OSF 注册表于 2023 年 11 月 10 日注册,https://osf.io/s2gct 。