Stenton Sarah L, O'Leary Melanie, Lemire Gabrielle, VanNoy Grace E, DiTroia Stephanie, Ganesh Vijay S, Groopman Emily, O'Heir Emily, Mangilog Brian, Osei-Owusu Ikeoluwa, Pais Lynn S, Serrano Jillian, Singer-Berk Moriel, Weisburd Ben, Wilson Michael, Austin-Tse Christina, Abdelhakim Marwa, Althagafi Azza, Babbi Giulia, Bellazzi Riccardo, Bovo Samuele, Carta Maria Giulia, Casadio Rita, Coenen Pieter-Jan, De Paoli Federica, Floris Matteo, Gajapathy Manavalan, Hoehndorf Robert, Jacobsen Julius O B, Joseph Thomas, Kamandula Akash, Katsonis Panagiotis, Kint Cyrielle, Lichtarge Olivier, Limongelli Ivan, Lu Yulan, Magni Paolo, Mamidi Tarun Karthik Kumar, Martelli Pier Luigi, Mulargia Marta, Nicora Giovanna, Nykamp Keith, Pejaver Vikas, Peng Yisu, Pham Thi Hong Cam, Podda Maurizio S, Rao Aditya, Rizzo Ettore, Saipradeep Vangala G, Savojardo Castrense, Schols Peter, Shen Yang, Sivadasan Naveen, Smedley Damian, Soru Dorian, Srinivasan Rajgopal, Sun Yuanfei, Sunderam Uma, Tan Wuwei, Tiwari Naina, Wang Xiao, Wang Yaqiong, Williams Amanda, Worthey Elizabeth A, Yin Rujie, You Yuning, Zeiberg Daniel, Zucca Susanna, Bakolitsa Constantina, Brenner Steven E, Fullerton Stephanie M, Radivojac Predrag, Rehm Heidi L, O'Donnell-Luria Anne
Division of Genetics and Genomics, Boston Children's Hospital, Harvard Medical School, Boston, MA, USA.
Program in Medical and Population Genetics, Broad Institute of MIT and Harvard, Cambridge, MA, USA.
medRxiv. 2023 Aug 4:2023.08.02.23293212. doi: 10.1101/2023.08.02.23293212.
A major obstacle faced by rare disease families is obtaining a genetic diagnosis. The average "diagnostic odyssey" lasts over five years, and causal variants are identified in under 50%. The Rare Genomes Project (RGP) is a direct-to-participant research study on the utility of genome sequencing (GS) for diagnosis and gene discovery. Families are consented for sharing of sequence and phenotype data with researchers, allowing development of a Critical Assessment of Genome Interpretation (CAGI) community challenge, placing variant prioritization models head-to-head in a real-life clinical diagnostic setting.
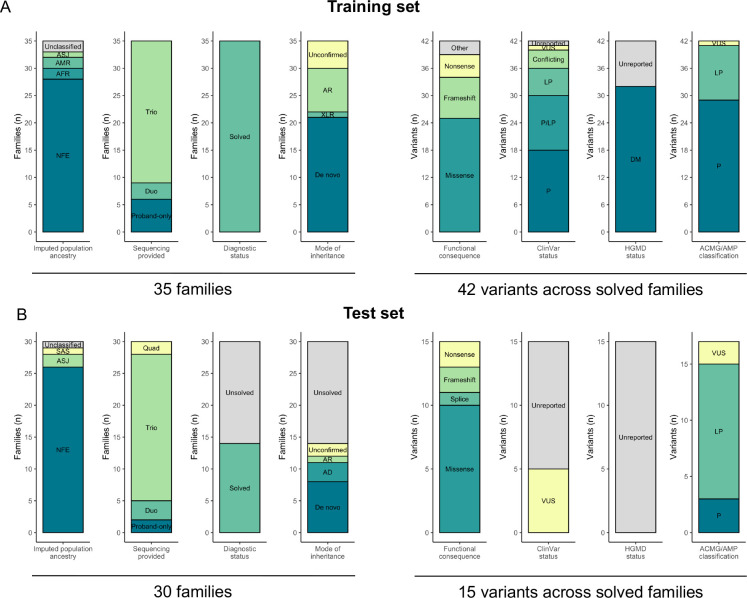
Predictors were provided a dataset of phenotype terms and variant calls from GS of 175 RGP individuals (65 families), including 35 solved training set families, with causal variants specified, and 30 test set families (14 solved, 16 unsolved). The challenge tasked teams with identifying the causal variants in as many test set families as possible. Ranked variant predictions were submitted with estimated probability of causal relationship (EPCR) values. Model performance was determined by two metrics, a weighted score based on rank position of true positive causal variants and maximum F-measure, based on precision and recall of causal variants across EPCR thresholds.
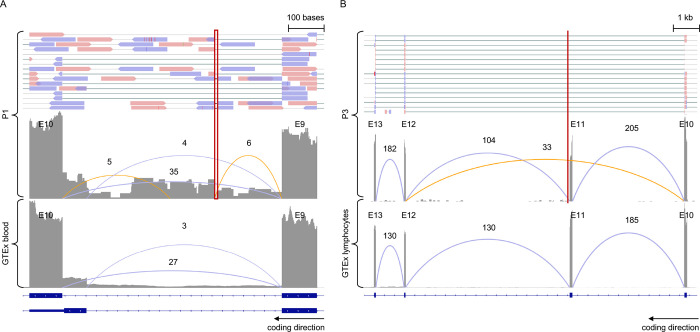
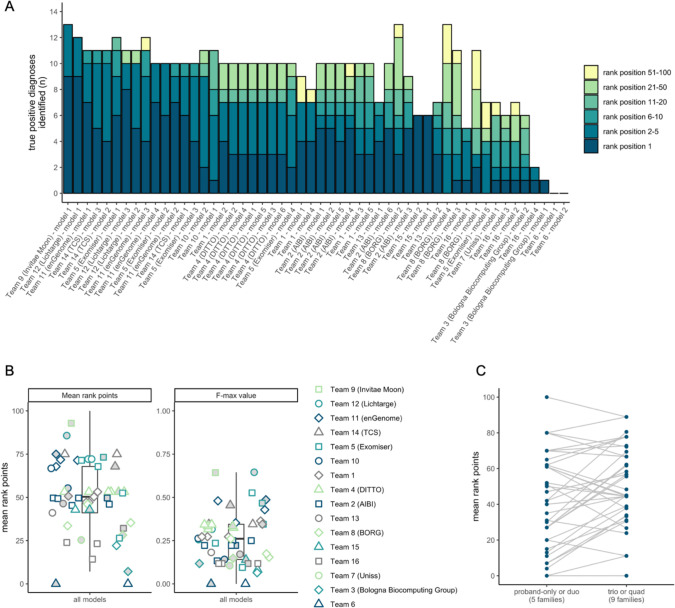
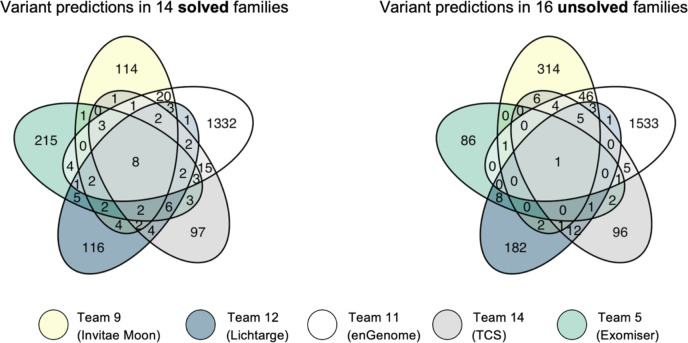
Sixteen teams submitted predictions from 52 models, some with manual review incorporated. Top performing teams recalled the causal variants in up to 13 of 14 solved families by prioritizing high quality variant calls that were rare, predicted deleterious, segregating correctly, and consistent with reported phenotype. In unsolved families, newly discovered diagnostic variants were returned to two families following confirmatory RNA sequencing, and two prioritized novel disease gene candidates were entered into Matchmaker Exchange. In one example, RNA sequencing demonstrated aberrant splicing due to a deep intronic indel in , identified in with a frameshift variant, in an unsolved proband with phenotype overlap with asparagine synthetase deficiency.
By objective assessment of variant predictions, we provide insights into current state-of-the-art algorithms and platforms for genome sequencing analysis for rare disease diagnosis and explore areas for future optimization. Identification of diagnostic variants in unsolved families promotes synergy between researchers with clinical and computational expertise as a means of advancing the field of clinical genome interpretation.
罕见病家庭面临的一个主要障碍是获得基因诊断。平均“诊断之旅”持续超过五年,且仅不到50%的家庭能确定致病变异。罕见基因组计划(RGP)是一项直接面向参与者的研究,旨在探讨基因组测序(GS)在诊断和基因发现方面的效用。研究征得家庭同意,让研究人员共享序列和表型数据,从而促成了基因组解读关键评估(CAGI)社区挑战赛,使变异优先级排序模型在真实临床诊断环境中进行直接较量。
向预测者提供了来自175名RGP个体(65个家庭)GS的表型术语和变异调用数据集,其中包括35个已解决的训练集家庭(已明确致病变异)以及30个测试集家庭(14个已解决,16个未解决)。挑战赛要求各团队尽可能多地识别测试集家庭中的致病变异。提交的变异预测排名需附带因果关系估计概率(EPCR)值。模型性能由两个指标确定,一个是基于真阳性致病变异排名位置的加权分数,另一个是基于EPCR阈值的致病变异精度和召回率的最大F值。
16个团队提交了来自52个模型的预测结果,有些模型还纳入了人工审核。表现最佳的团队通过优先考虑罕见、预测为有害、正确分离且与报告表型一致的高质量变异调用,在14个已解决家庭中的多达13个家庭中识别出致病变异。在未解决的家庭中,经确认性RNA测序后,两个家庭发现了新的诊断变异,并且两个优先级较高的新型疾病基因候选物被录入匹配者交换平台。在一个例子中,RNA测序显示,在一名与天冬酰胺合成酶缺乏症表型重叠的未解决先证者中,由于在 中一个内含子深处的插入缺失导致异常剪接,该插入缺失与一个移码变异一起被识别出来。
通过对变异预测的客观评估,我们深入了解了当前用于罕见病诊断的基因组测序分析的先进算法和平台,并探索了未来优化的领域。在未解决的家庭中识别诊断变异促进了具有临床和计算专业知识的研究人员之间的协同合作,以此推动临床基因组解读领域的发展。