Institute for Cellular and Integrative Physiology, Center for Experimental Medicine, University Medical Center Hamburg-Eppendorf, 20246 Hamburg, Germany.
Institute of Human Genetics, University Medical Center Hamburg-Eppendorf, 20246 Hamburg, Germany.
Am J Hum Genet. 2024 Jun 6;111(6):1206-1221. doi: 10.1016/j.ajhg.2024.04.019. Epub 2024 May 20.
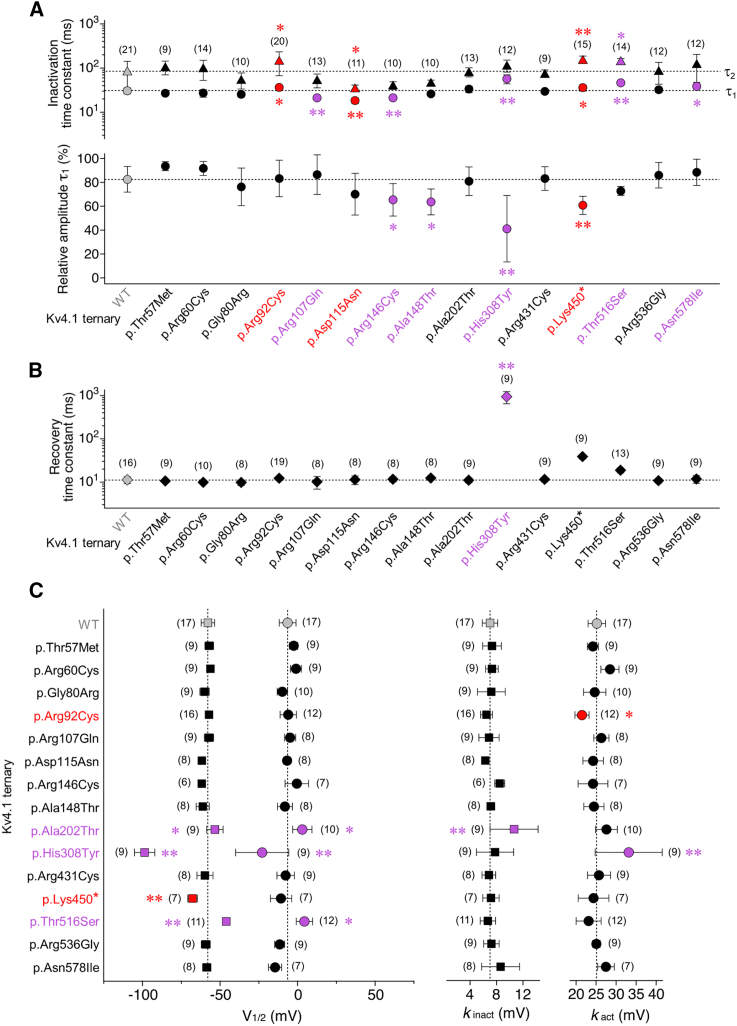
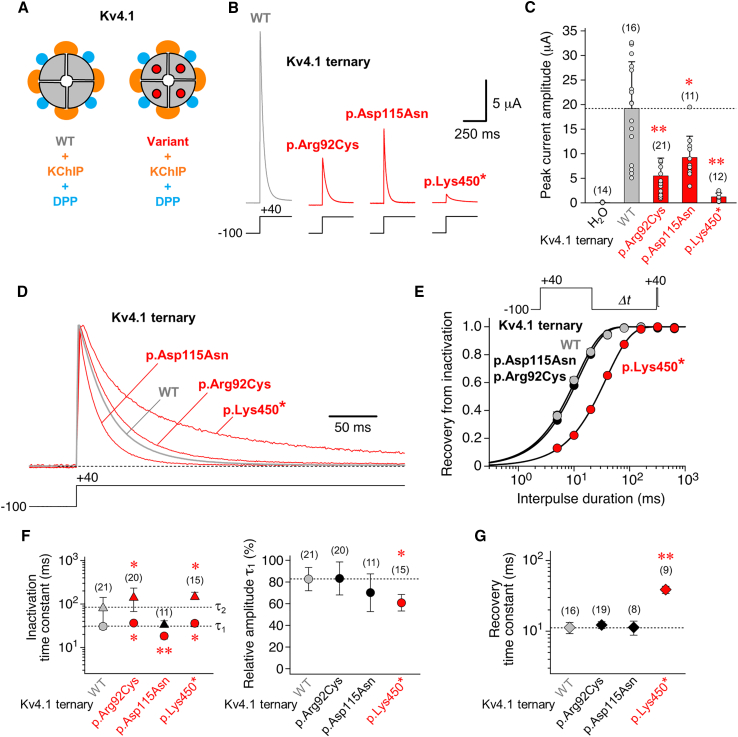
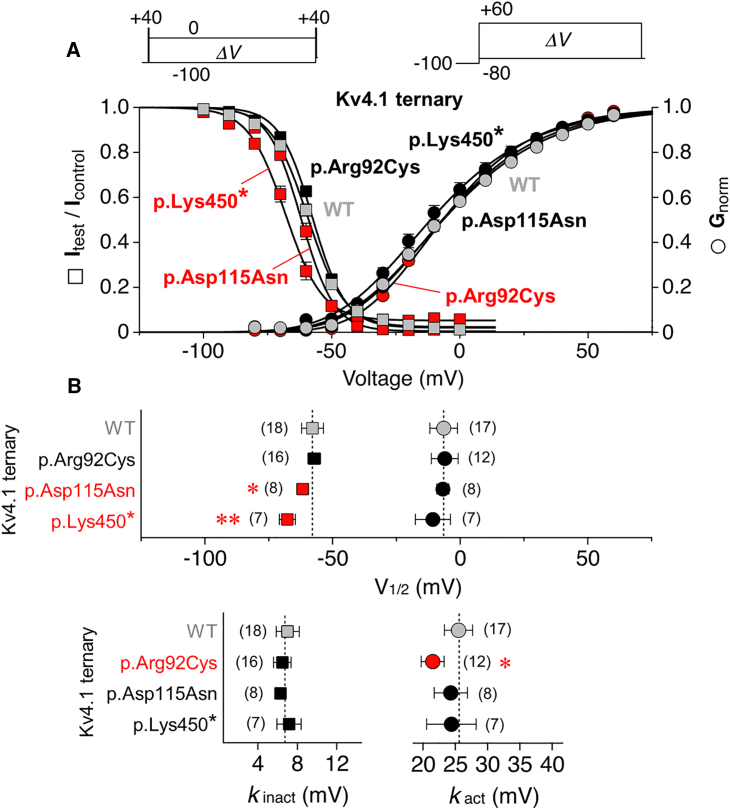
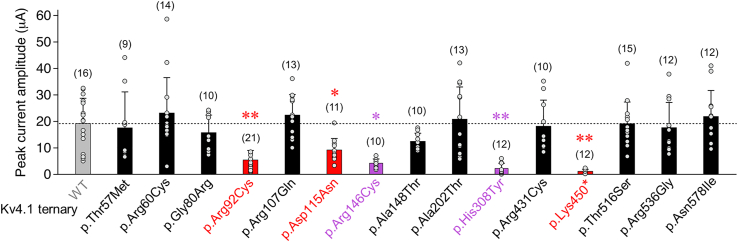
Utilizing trio whole-exome sequencing and a gene matching approach, we identified a cohort of 18 male individuals from 17 families with hemizygous variants in KCND1, including two de novo missense variants, three maternally inherited protein-truncating variants, and 12 maternally inherited missense variants. Affected subjects present with a neurodevelopmental disorder characterized by diverse neurological abnormalities, mostly delays in different developmental domains, but also distinct neuropsychiatric signs and epilepsy. Heterozygous carrier mothers are clinically unaffected. KCND1 encodes the α-subunit of Kv4.1 voltage-gated potassium channels. All variant-associated amino acid substitutions affect either the cytoplasmic N- or C-terminus of the channel protein except for two occurring in transmembrane segments 1 and 4. Kv4.1 channels were functionally characterized in the absence and presence of auxiliary β subunits. Variant-specific alterations of biophysical channel properties were diverse and varied in magnitude. Genetic data analysis in combination with our functional assessment shows that Kv4.1 channel dysfunction is involved in the pathogenesis of an X-linked neurodevelopmental disorder frequently associated with a variable neuropsychiatric clinical phenotype.
利用 trio 全外显子测序和基因匹配方法,我们从 17 个家系中鉴定了 18 名男性个体,他们携带 KCND1 的半合子变异,包括两个新生错义变异、三个母系遗传的蛋白截断变异和 12 个母系遗传的错义变异。受影响的个体表现出一种神经发育障碍,其特征是多种神经异常,主要是不同发育领域的延迟,但也有明显的神经精神症状和癫痫。杂合子携带者母亲无临床症状。KCND1 编码 Kv4.1 电压门控钾通道的 α 亚单位。所有与变异相关的氨基酸取代都影响通道蛋白的胞质 N 端或 C 端,除了两个发生在跨膜片段 1 和 4 中。在不存在和存在辅助 β 亚单位的情况下,对 Kv4.1 通道的功能进行了特征描述。变异特异性的生物物理通道特性改变是多样的,幅度也不同。遗传数据分析结合我们的功能评估表明,Kv4.1 通道功能障碍与一种常见的伴有可变神经精神临床表型的 X 连锁神经发育障碍的发病机制有关。