Department of Computer Science, University of Kentucky, 329 Rose St, Lexington, KY, 40508, USA.
Department of Plant Pathology, University of Kentucky, 1405 Veterans Dr, Lexington, KY, 40546, USA.
BMC Bioinformatics. 2024 Jun 4;25(1):205. doi: 10.1186/s12859-024-05811-9.
Although RNA-seq data are traditionally used for quantifying gene expression levels, the same data could be useful in an integrated approach to compute genetic distances as well. Challenges to using mRNA sequences for computing genetic distances include the relatively high conservation of coding sequences and the presence of paralogous and, in some species, homeologous genes.
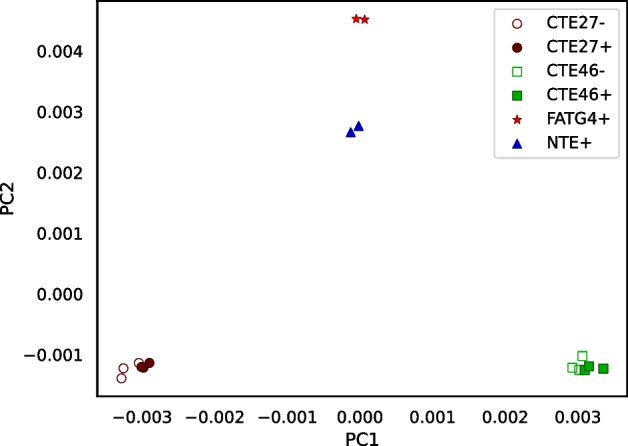
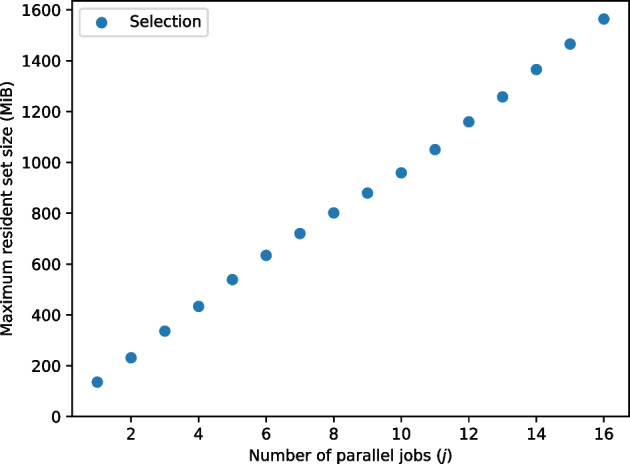
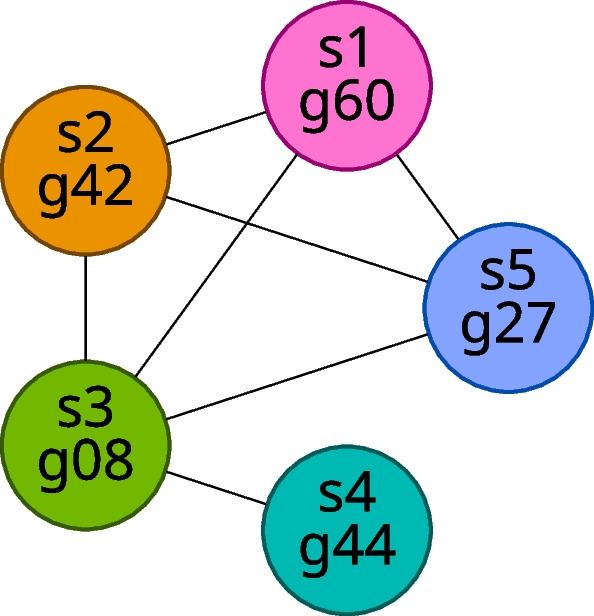
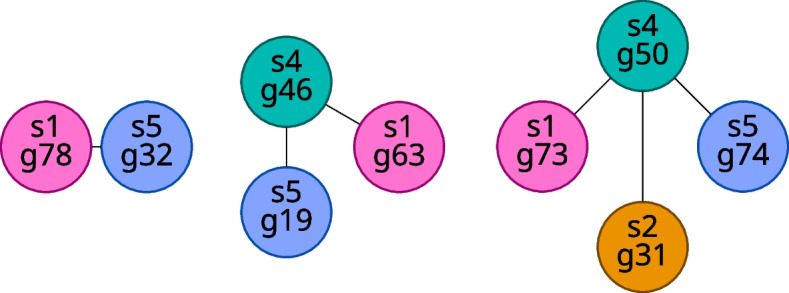
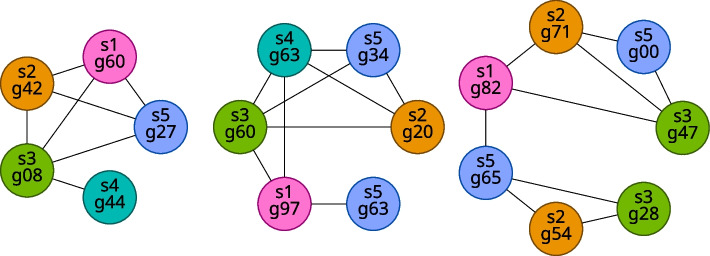
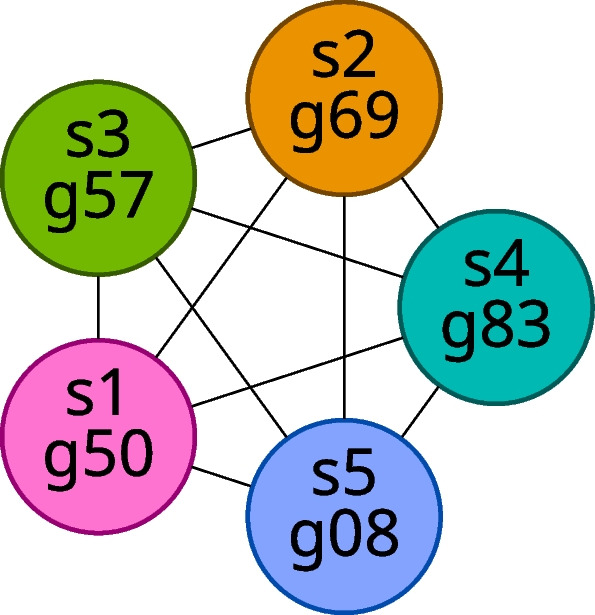
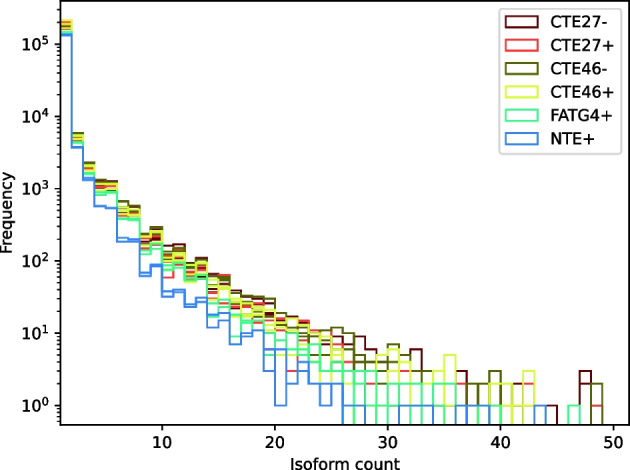
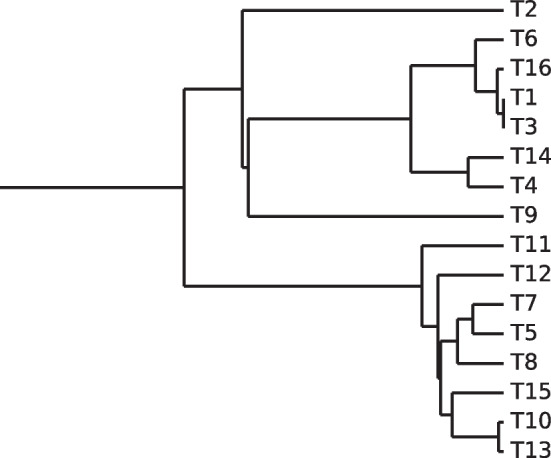
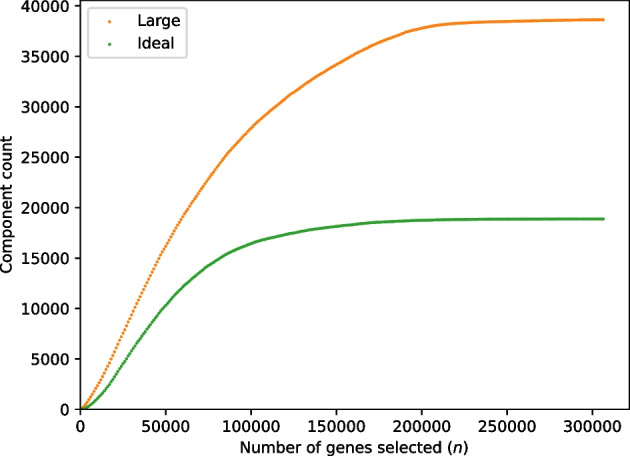
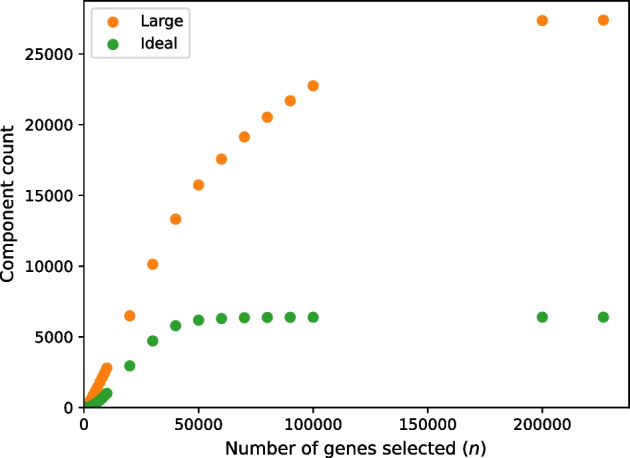
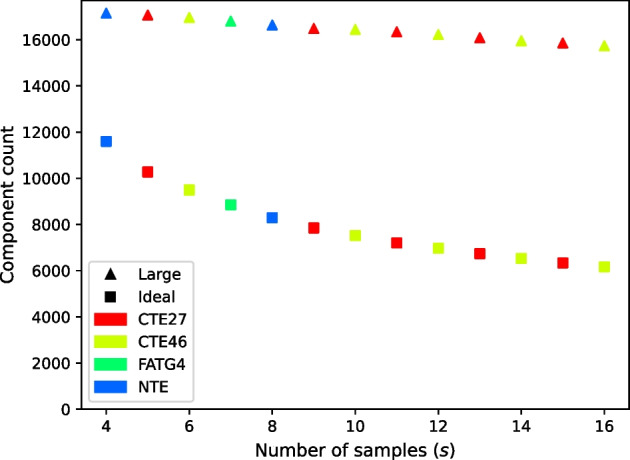
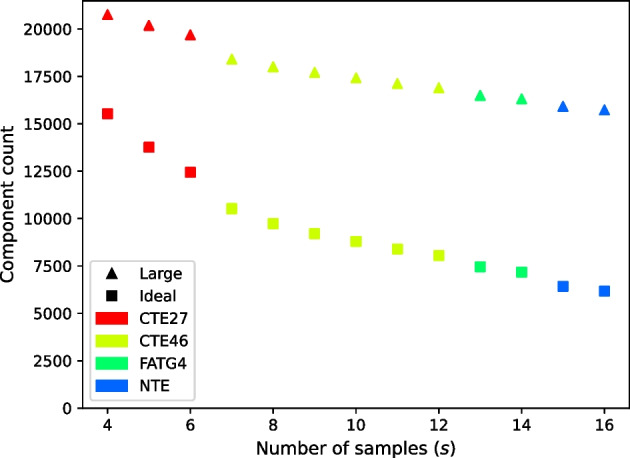
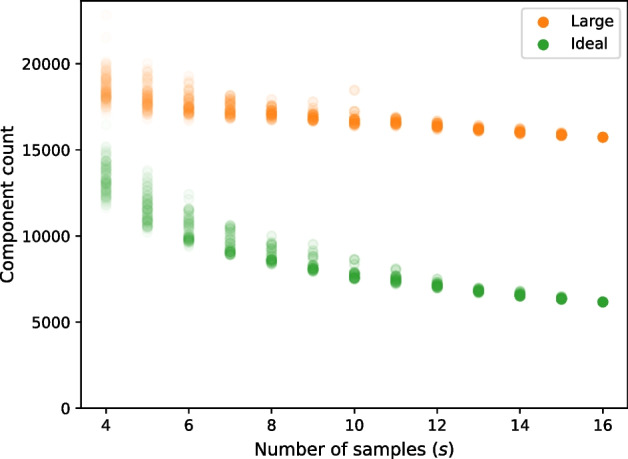
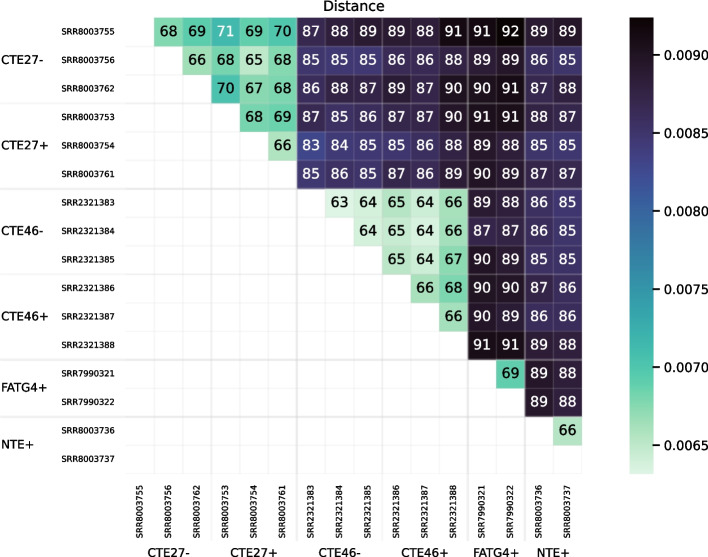
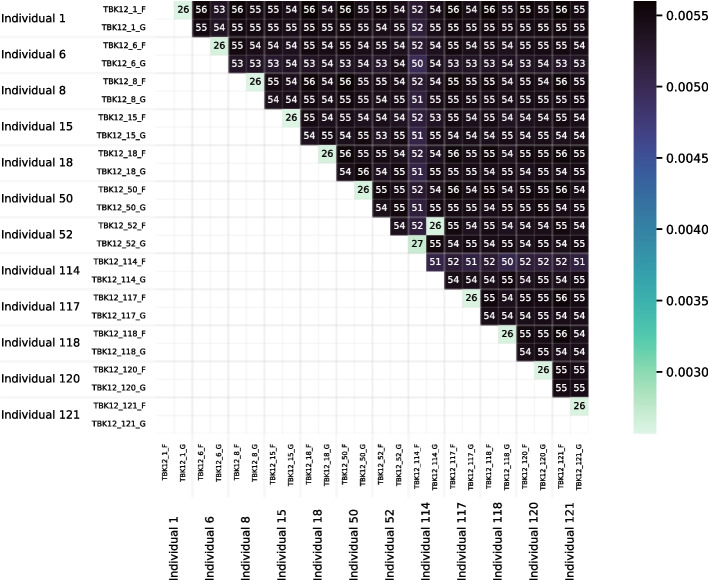
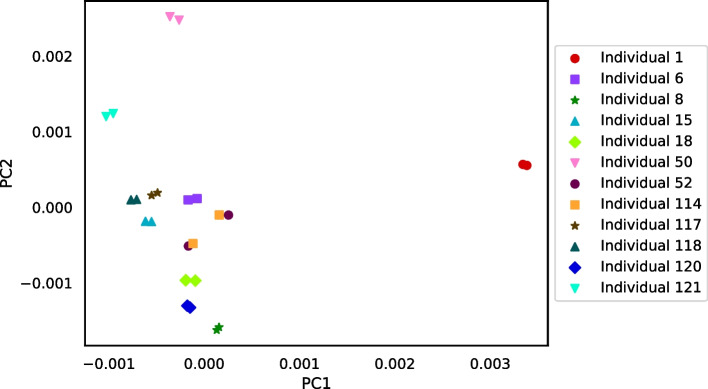
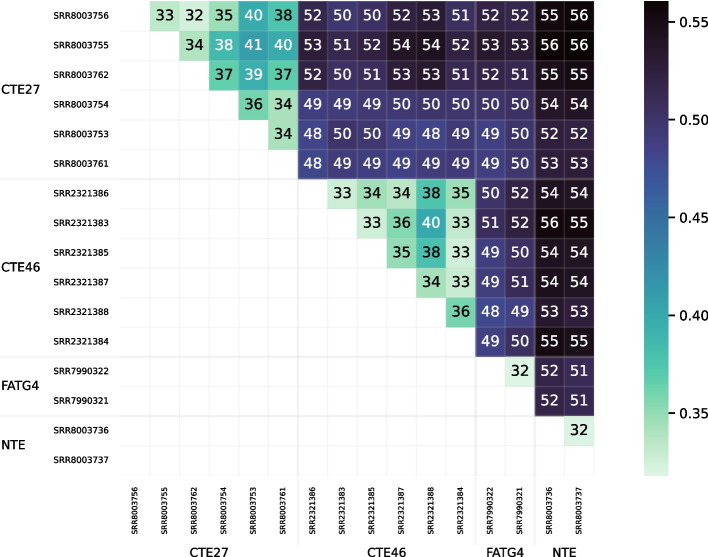
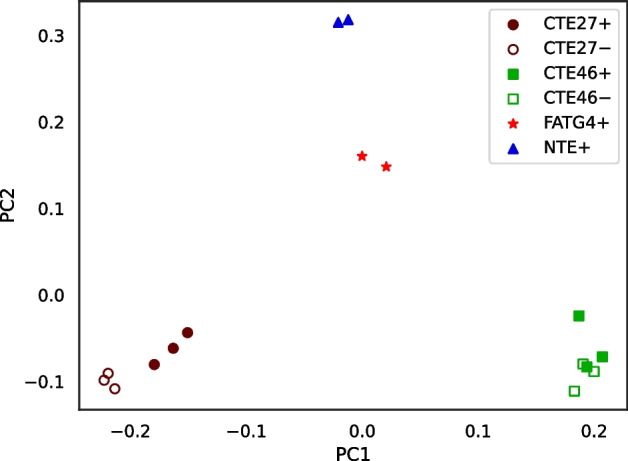
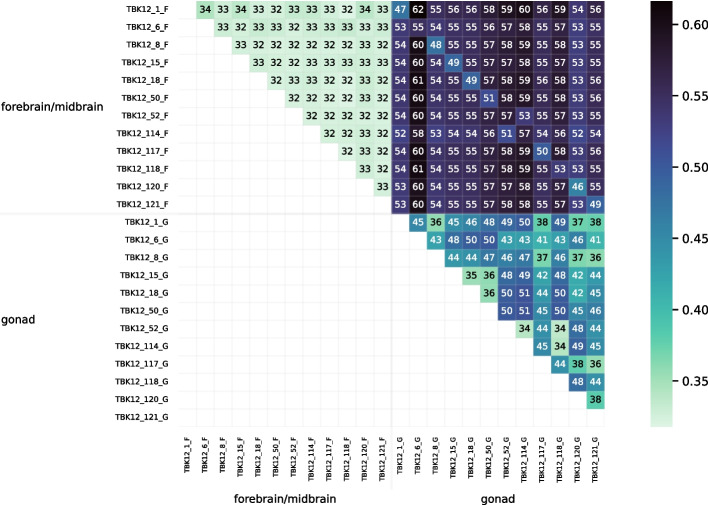
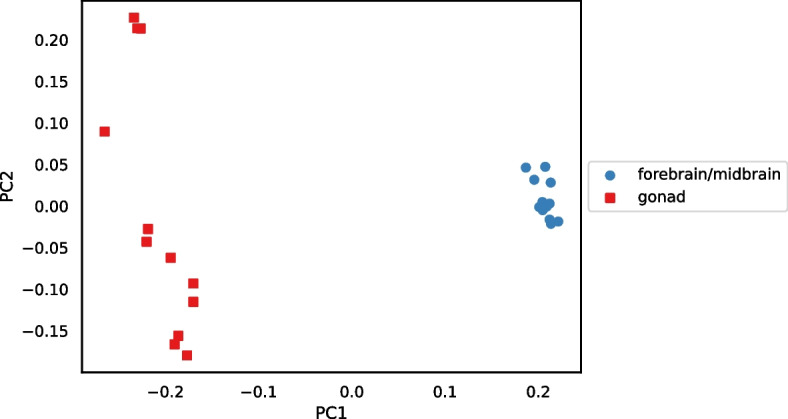
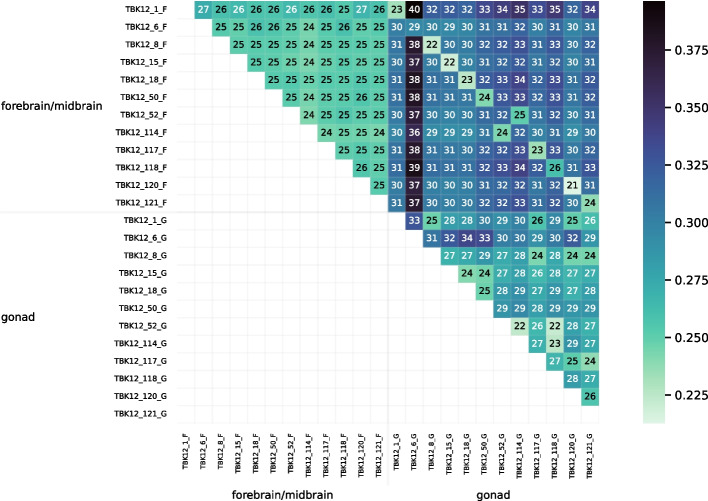
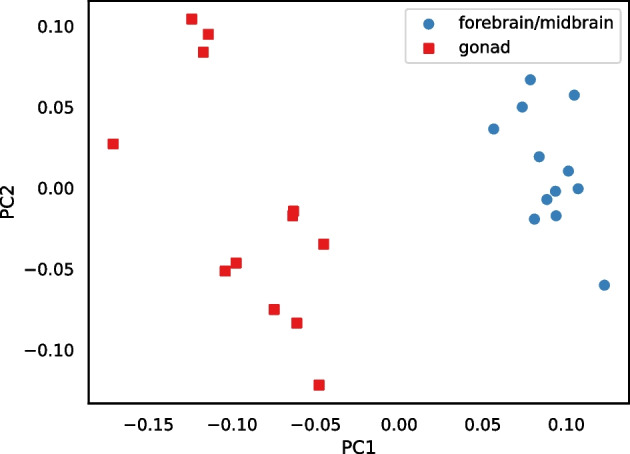
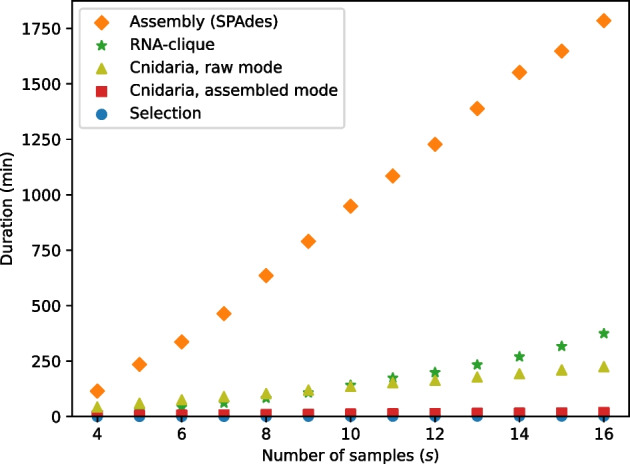
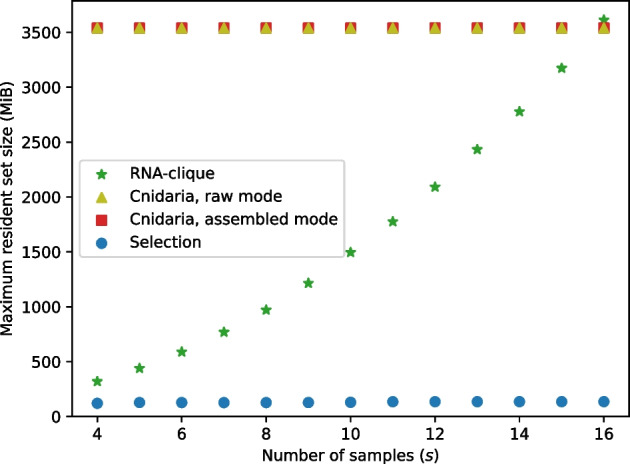
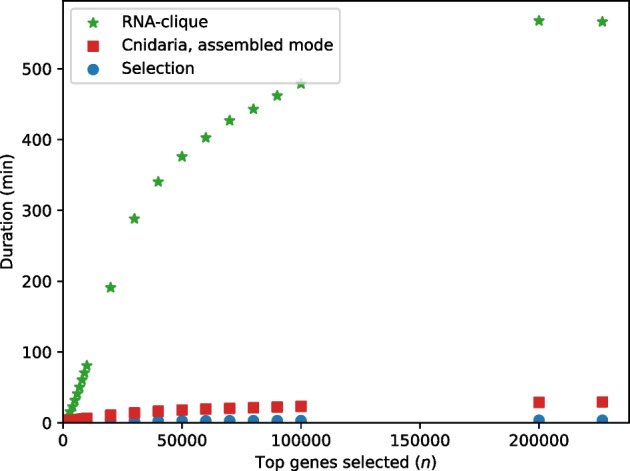
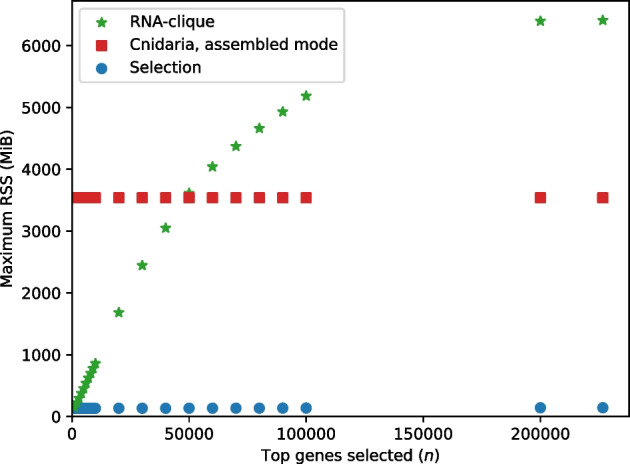
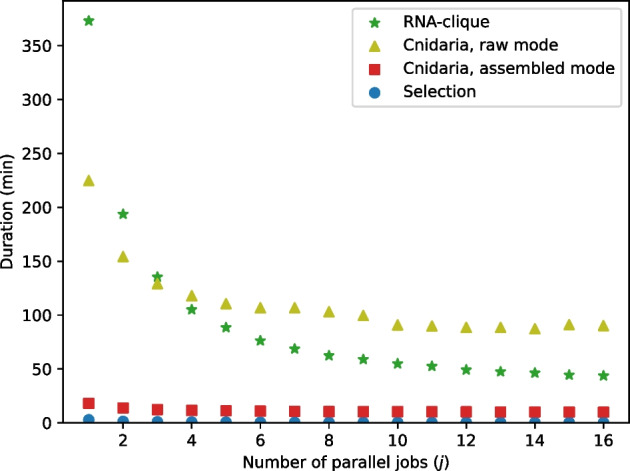
We developed a new computational method, RNA-clique, for calculating genetic distances using assembled RNA-seq data and assessed the efficacy of the method using biological and simulated data. The method employs reciprocal BLASTn followed by graph-based filtering to ensure that only orthologous genes are compared. Each vertex in the graph constructed for filtering represents a gene in a specific sample under comparison, and an edge connects a pair of vertices if the genes they represent are best matches for each other in their respective samples. The distance computation is a function of the BLAST alignment statistics and the constructed graph and incorporates only those genes that are present in some complete connected component of this graph. As a biological testbed we used RNA-seq data of tall fescue (Lolium arundinaceum), an allohexaploid plant ( ), and bluehead wrasse (Thalassoma bifasciatum), a teleost fish. RNA-clique reliably distinguished individual tall fescue plants by genotype and distinguished bluehead wrasse RNA-seq samples by individual. In tests with simulated RNA-seq data, the ground truth phylogeny was accurately recovered from the computed distances. Moreover, tests of the algorithm parameters indicated that, even with stringent filtering for orthologs, sufficient sequence data were retained for the distance computations. Although comparisons with an alternative method revealed that RNA-clique has relatively high time and memory requirements, the comparisons also showed that RNA-clique's results were at least as reliable as the alternative's for tall fescue data and were much more reliable for the bluehead wrasse data.
Results of this work indicate that RNA-clique works well as a way of deriving genetic distances from RNA-seq data, thus providing a methodological integration of functional and genetic diversity studies.
尽管 RNA-seq 数据传统上用于量化基因表达水平,但相同的数据也可以在综合方法中用于计算遗传距离。使用 mRNA 序列计算遗传距离的挑战包括编码序列的相对高保守性以及同源和基因的存在。
我们开发了一种新的计算方法 RNA-clique,用于使用组装的 RNA-seq 数据计算遗传距离,并使用生物和模拟数据评估该方法的功效。该方法采用相互 BLASTn 随后进行基于图的过滤,以确保仅比较同源基因。用于过滤构建的图中的每个顶点代表比较中特定样本中的基因,如果它们所代表的基因在各自的样本中彼此是最佳匹配,则顶点之间存在边缘。距离计算是 BLAST 对齐统计数据和构建图的函数,并仅包含该图的某些完整连通分量中存在的那些基因。作为生物学测试平台,我们使用了 tall fescue(Lolium arundinaceum)的 RNA-seq 数据,该数据为异源六倍体植物( ),以及蓝头濑鱼(Thalassoma bifasciatum)的 RNA-seq 数据,该数据为硬骨鱼。RNA-clique 可靠地根据基因型区分 tall fescue 植物,并且根据个体区分蓝头濑鱼 RNA-seq 样本。在使用模拟 RNA-seq 数据的测试中,从计算的距离中准确恢复了实际的系统发育。此外,对算法参数的测试表明,即使对同源物进行严格过滤,也保留了足够的序列数据用于距离计算。尽管与替代方法的比较表明 RNA-clique 具有相对较高的时间和内存要求,但比较还表明,对于 tall fescue 数据,RNA-clique 的结果至少与替代方法一样可靠,而对于蓝头濑鱼数据,则要可靠得多。
这项工作的结果表明,RNA-clique 是从 RNA-seq 数据中获取遗传距离的有效方法,从而为功能和遗传多样性研究提供了方法上的整合。