University of Utah Molecular Medicine Program, Salt Lake City, Utah, USA; Department of Emergency Medicine Washington University School, St. Louis, Missouri, USA.
University of Utah Molecular Medicine Program, Salt Lake City, Utah, USA.
J Thromb Haemost. 2024 Sep;22(9):2576-2588. doi: 10.1016/j.jtha.2024.05.025. Epub 2024 Jun 6.
Aging is an independent risk factor for the development of cardiovascular, thrombotic, and other chronic diseases. However, mechanisms of platelet hyperactivation in aging remain poorly understood.
Here, we examine whether and how aging alters intracellular signaling in platelets to support platelet hyperactivity and thrombosis.
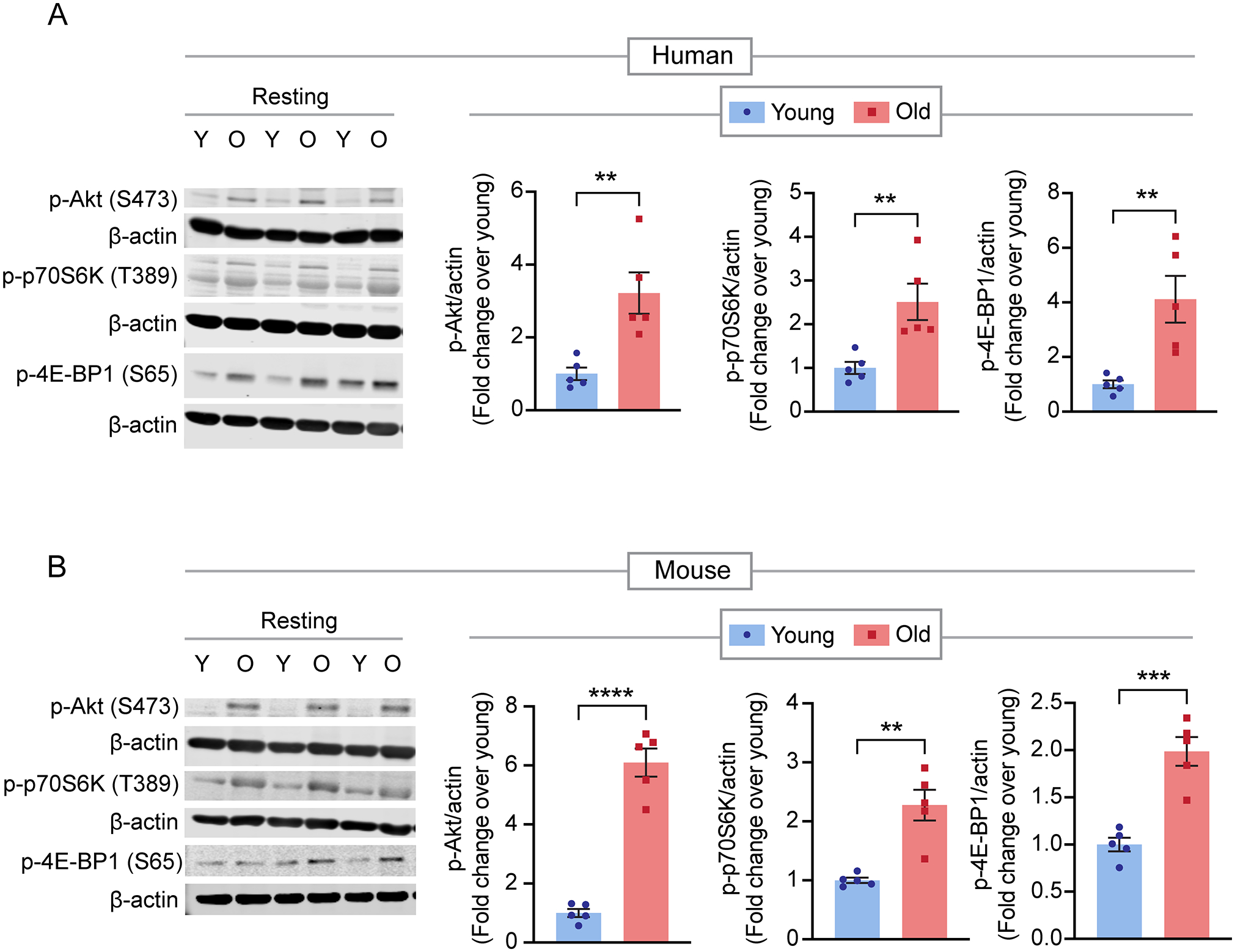
Quantitative mass spectrometry with tandem mass tag labeling systematically measured protein phosphorylation in platelets from healthy aged (>65 years) and young human (<45 years) subjects. The role of platelet mechanistic target of rapamycin (mTOR) in aging-induced platelet hyperreactivity was assessed using pharmacologic mTOR inhibition and a platelet-specific mTOR-deficient mouse model (mTOR).
Quantitative phosphoproteomics uncovered differential site-specific protein phosphorylation within mTOR, Rho GTPase, and MAPK pathways in platelets from aged donors. Western blot confirmed constitutive activation of the mTOR pathway in platelets from both aged humans and mice, which was associated with increased aggregation compared with that in young controls. Inhibition of mTOR with either Torin 1 in aged humans or genetic deletion in aged mice reversed platelet hyperreactivity. In a collagen-epinephrine pulmonary thrombosis model, aged wild-type (mTOR) mice succumbed significantly faster than young controls, while time to death of aged mTOR mice was similar to that of young mTOR mice. Mechanistically, we noted increased Rac1 activation and levels of mitochondrial reactive oxygen species in resting platelets from aged mice, as well as increased p38 phosphorylation upstream of thromboxane generation following agonist stimulation.
Aging-related changes in mTOR phosphorylation enhance Rac1 and p38 activation to enhance thromboxane generation, platelet hyperactivity, and thrombosis.
衰老本身是心血管、血栓形成和其他慢性疾病发展的一个独立危险因素。然而,衰老导致血小板过度激活的机制仍知之甚少。
本研究旨在探讨衰老是否以及如何改变血小板内信号转导,以支持血小板过度激活和血栓形成。
采用串联质量标签标记的定量质谱法系统检测来自健康老年(>65 岁)和年轻(<45 岁)个体的血小板中的蛋白磷酸化。使用药理学 mTOR 抑制和血小板特异性 mTOR 缺陷小鼠模型(mTOR)评估血小板机械性靶标雷帕霉素(mTOR)在衰老诱导的血小板高反应性中的作用。
定量磷酸化蛋白质组学揭示了 mTOR、Rho GTPase 和 MAPK 途径中特定部位的蛋白磷酸化在老年供体血小板中的差异。Western blot 证实了来自老年人和小鼠的血小板中 mTOR 通路的组成性激活,与年轻对照组相比,其聚集性增加。在胶原-肾上腺素肺血栓形成模型中,老年野生型(mTOR)小鼠的死亡率明显高于年轻对照组,而老年 mTOR 小鼠的死亡率与年轻 mTOR 小鼠相似。在机制上,我们注意到老年小鼠静息血小板中 Rac1 的激活和线粒体活性氧水平增加,以及激动剂刺激后血栓烷生成上游 p38 磷酸化增加。
mTOR 磷酸化的衰老相关变化增强了 Rac1 和 p38 的激活,从而增强了血栓烷生成、血小板过度激活和血栓形成。