Voss Katalin, Kaur Katrina M, Banerjee Rituparna, Breden Felix, Pennell Matt
Department of Quantitative and Computational Biology, University of Southern California, USA.
Department of Zoology, University of British Columbia, Canada.
bioRxiv. 2024 Jun 2:2024.05.29.596491. doi: 10.1101/2024.05.29.596491.
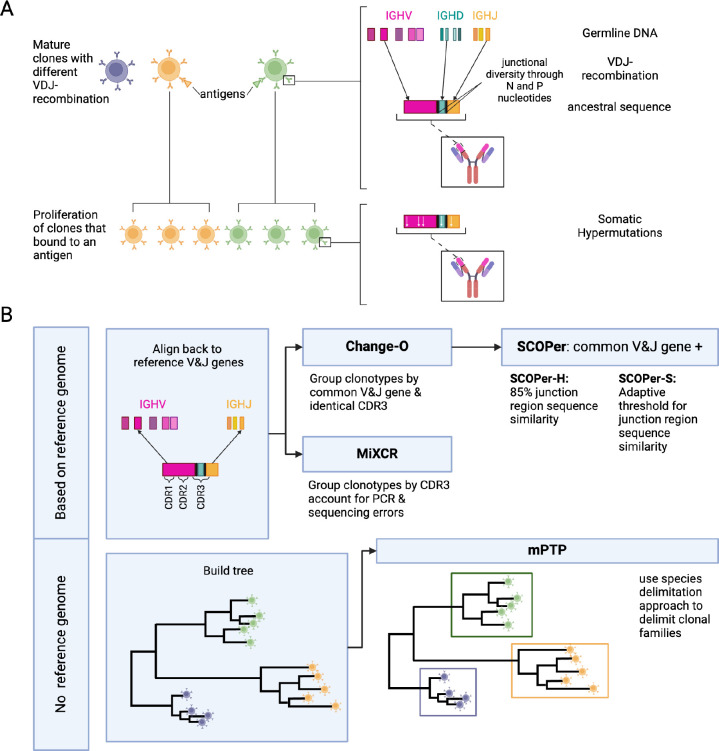
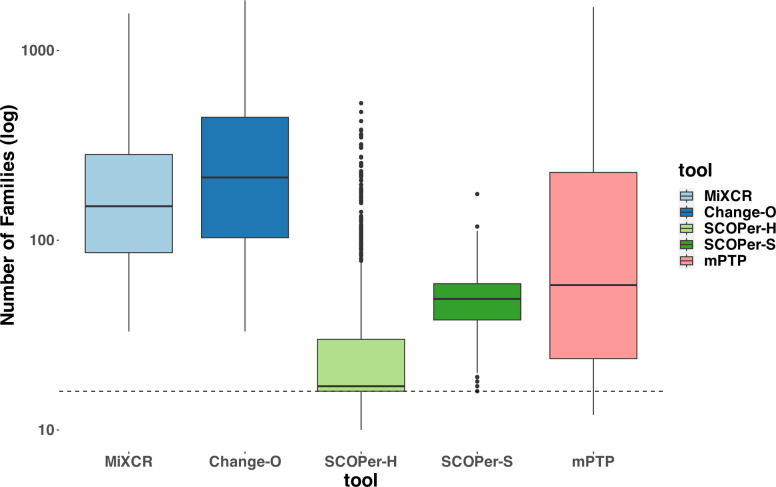
The adaptive immune response relies on a diverse repertoire of B-cell receptors, each of which is characterized by a distinct sequence resulting from VDJ-recombination. Upon binding to an antigen, B-cells undergo clonal expansion and in a process unique to B-cells the overall binding affinity of the repertoire is further enhanced by somatic hypermutations in the receptor sequence. For B-cell repertoires it is therefore particularly important to analyze the dynamics of clonal expansion and patterns of somatic hypermutations and thus it is necessary to group the sequences into distinct clones to determine the number and identity of expanding clonal families responding to an antigen. Multiple methods are currently used to identify clones from sequences, employing distinct approaches to the problem. Until now there has not been an extensive comparison of how well these methods perform under the same conditions. Furthermore, since this is fundamentally a phylogenetics problem, we speculated that the mPTP method, which delimits species based on an analysis of changes in the underlying process of diversification, might perform as well as or better than existing methods. Here we conducted extensive simulations of B-cell repertoires under a diverse set of conditions and studied errors in clonal assignment and in downstream ancestral state reconstruction. We demonstrated that SCOPer-H consistently yielded superior results across parameters. However, this approach relies on a good reference assembly for the germline immunoglobulin genes which is lacking for many species. Using mPTP had lower error rates than tailor-made immunogenetic methods and should therefore be considered by researchers studying antibody evolution in non-model organisms without a reference genome.
适应性免疫反应依赖于多样化的B细胞受体库,每个受体的特征是由VDJ重排产生的独特序列。与抗原结合后,B细胞经历克隆扩增,并且在B细胞特有的过程中,受体序列中的体细胞超突变进一步增强了受体库的整体结合亲和力。因此,对于B细胞受体库来说,分析克隆扩增的动态和体细胞超突变的模式尤为重要,因此有必要将序列分组为不同的克隆,以确定对抗原作出反应的扩增克隆家族的数量和身份。目前有多种方法用于从序列中识别克隆,采用了不同的方法来解决这个问题。到目前为止,还没有对这些方法在相同条件下的表现进行广泛的比较。此外,由于这从根本上是一个系统发育问题,我们推测基于对多样化潜在过程变化的分析来界定物种的mPTP方法可能与现有方法表现相当或更好。在这里,我们在各种条件下对B细胞受体库进行了广泛的模拟,并研究了克隆分配和下游祖先状态重建中的错误。我们证明,SCOPer-H在各个参数上始终产生优异的结果。然而,这种方法依赖于种系免疫球蛋白基因的良好参考组装,而许多物种都缺乏这种组装。使用mPTP的错误率低于特制的免疫遗传学方法,因此,对于在没有参考基因组的非模式生物中研究抗体进化的研究人员来说,应该考虑使用mPTP。