Shu Jirong, Wang Yuwei, Guo Weijie, Liu Tao, Cai Song, Shi Taoda, Hu Wenhao
School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou, 510006, China.
Shenzhen University Health Science Center, Shenzhen, 518060, China.
Commun Chem. 2024 Jun 12;7(1):135. doi: 10.1038/s42004-024-01213-3.
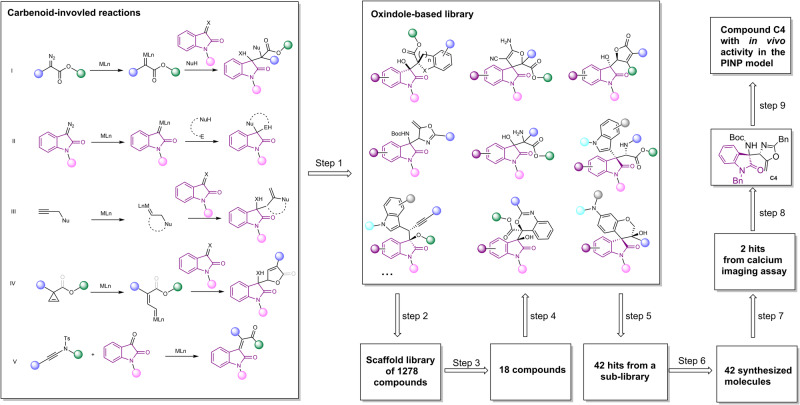
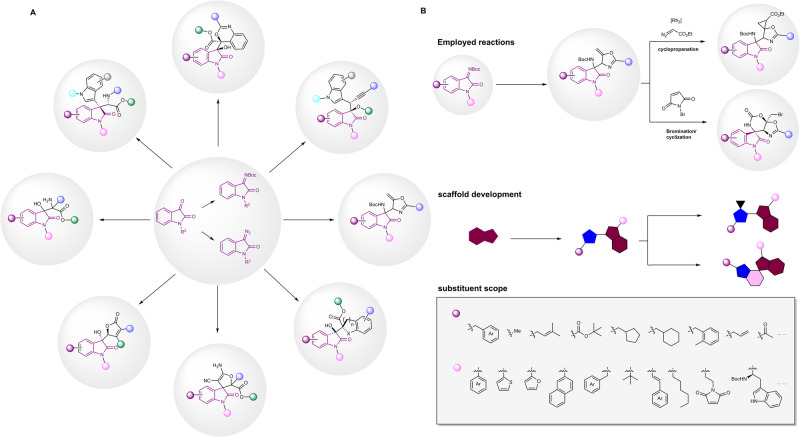
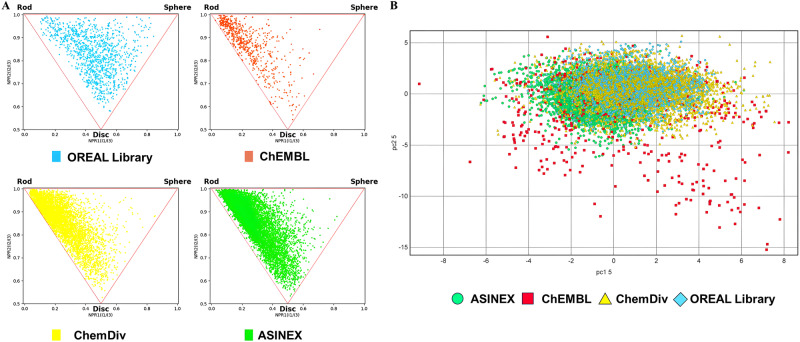
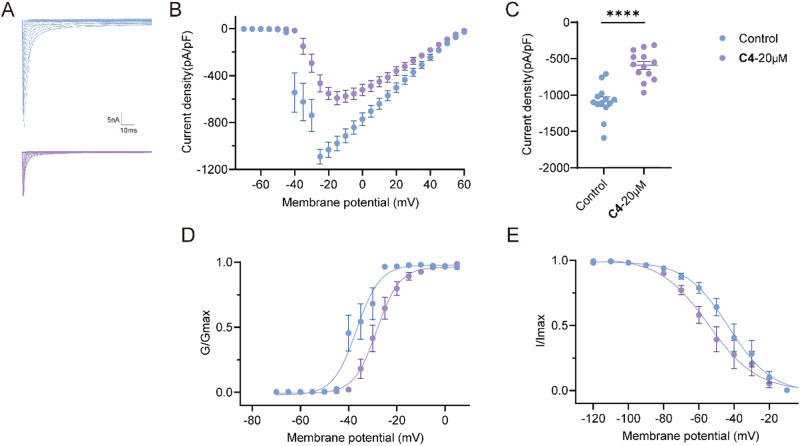
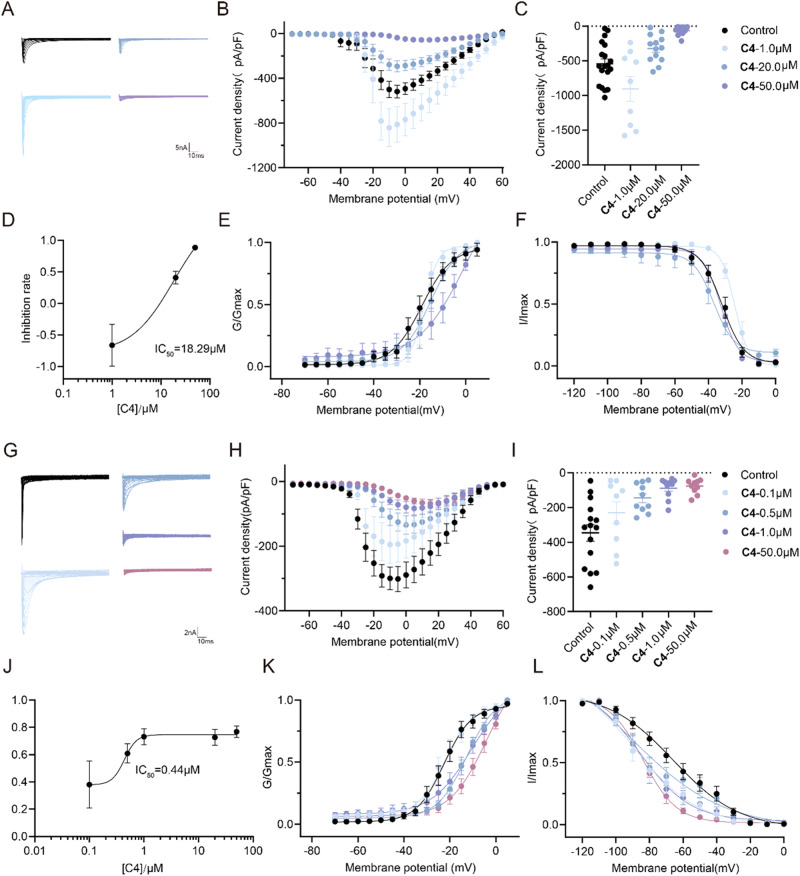
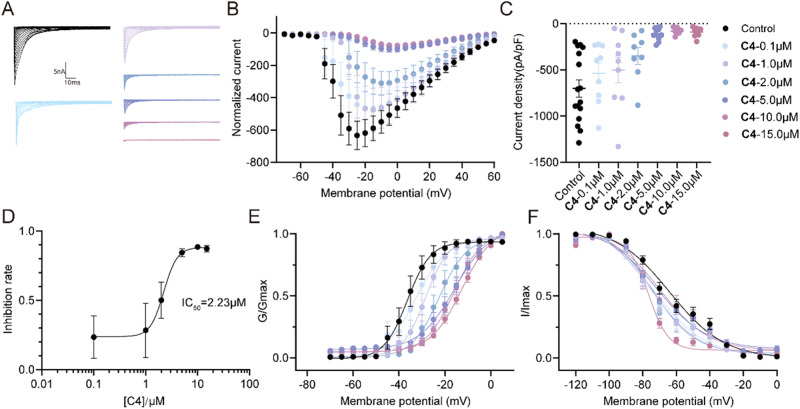
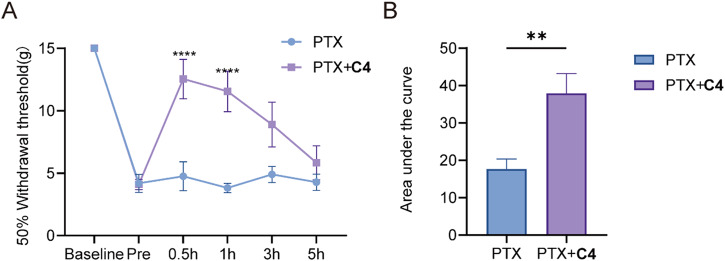
The discovery of selective Nav1.7 inhibitors is a promising approach for developing anti-nociceptive drugs. In this study, we present a novel oxindole-based readily accessible library (OREAL), which is characterized by readily accessibility, unique chemical space, ideal drug-like properties, and structural diversity. We used a scaffold-based approach to screen the OREAL and discovered compound C4 as a potent Nav1.7 inhibitor. The bioactivity characterization of C4 reveals that it is a selective Nav1.7 inhibitor and effectively reverses Paclitaxel-induced neuropathic pain (PINP) in rodent models. Preliminary toxicology study shows C4 is negative to hERG. The consistent results of molecular docking and molecular simulations further support the reasonability of the in-silico screening and show the insight of the binding mode of C4. Our discovery of C4 paves the way for pushing the Nav1.7-based anti-nociceptive drugs forward to the clinic.
发现选择性Nav1.7抑制剂是开发抗伤害感受药物的一种有前景的方法。在本研究中,我们展示了一种新型的基于氧化吲哚的易于获取的文库(OREAL),其特点是易于获取、独特的化学空间、理想的类药性质和结构多样性。我们采用基于骨架的方法筛选OREAL,并发现化合物C4是一种有效的Nav1.7抑制剂。C4的生物活性表征表明它是一种选择性Nav1.7抑制剂,并能有效逆转啮齿动物模型中紫杉醇诱导的神经性疼痛(PINP)。初步毒理学研究表明C4对人醚-去极化激活的钾离子通道(hERG)呈阴性。分子对接和分子模拟的一致结果进一步支持了虚拟筛选的合理性,并展示了C4结合模式的见解。我们对C4的发现为推动基于Nav1.7的抗伤害感受药物进入临床铺平了道路。