The Biodesign Institute, Arizona State University, Tempe, AZ, USA.
Ira A. Fulton Schools of Engineering, Arizona State University, Tempe, AZ, USA.
Mol Biol Evol. 2024 Jul 3;41(7). doi: 10.1093/molbev/msae117.
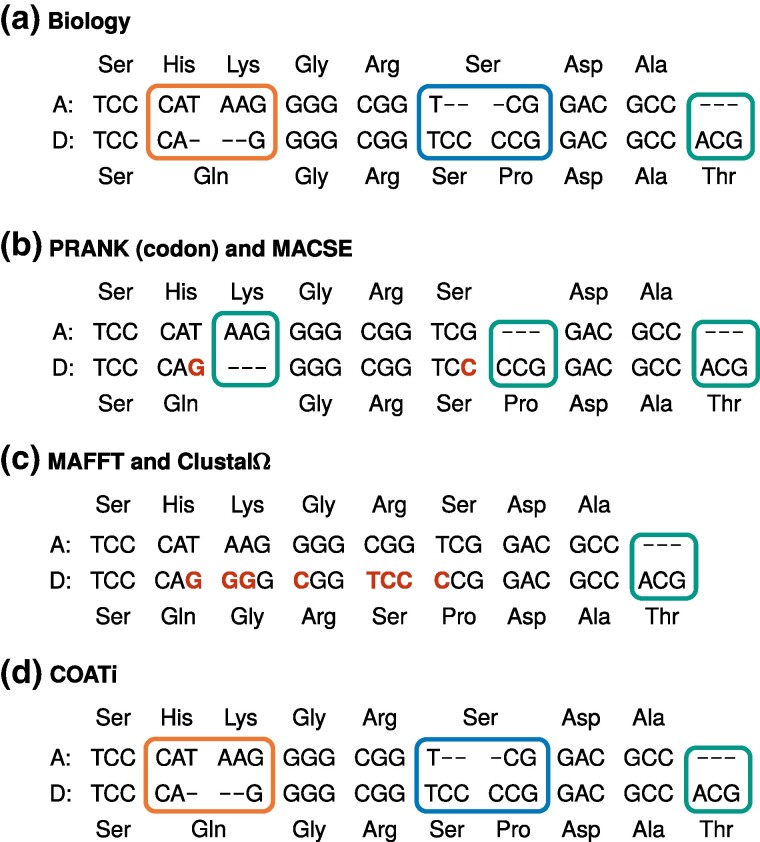
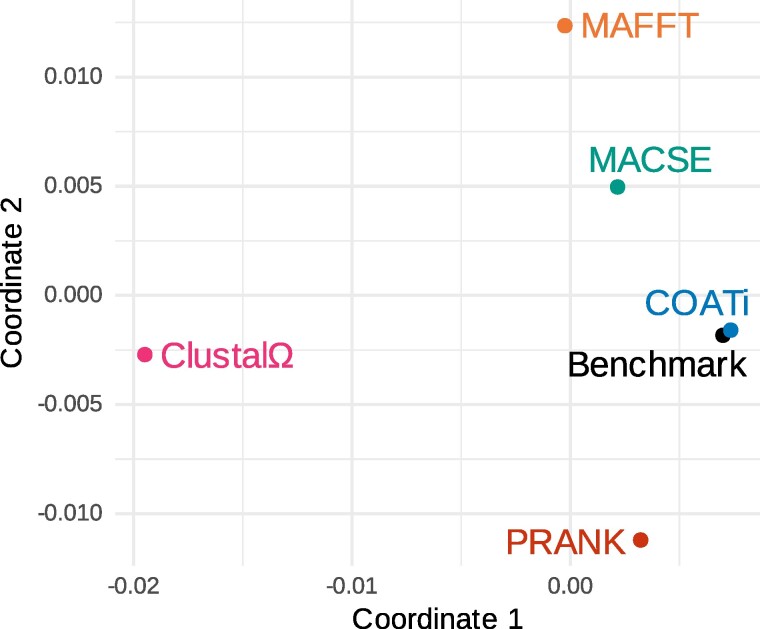
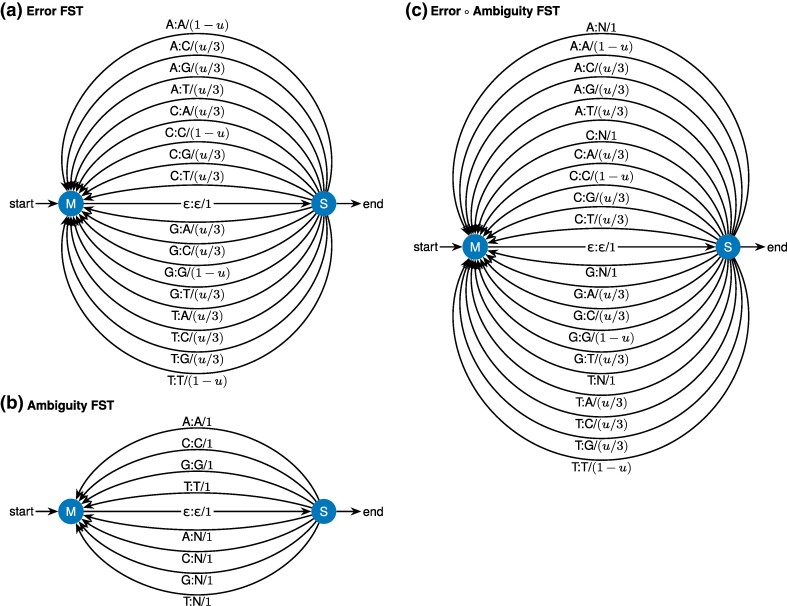
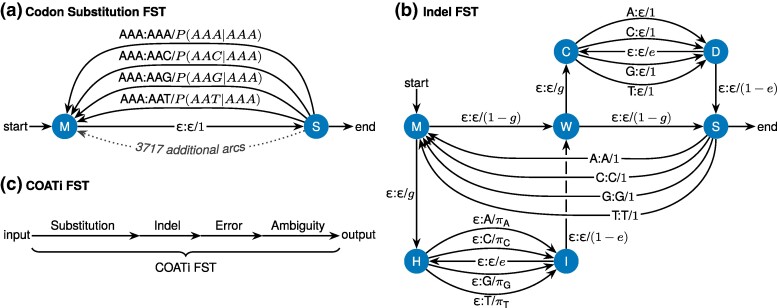
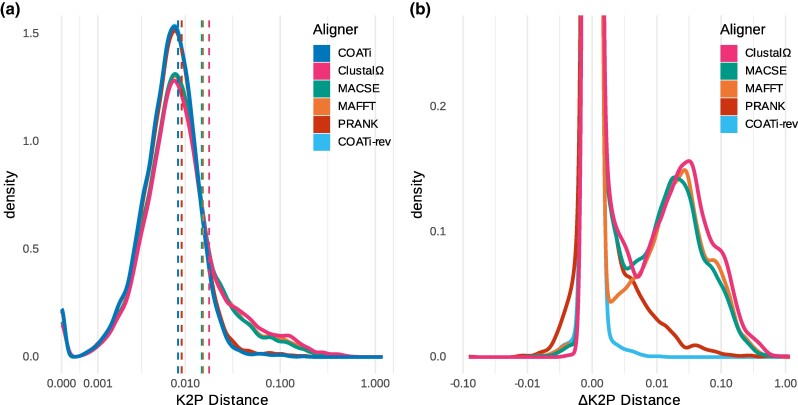
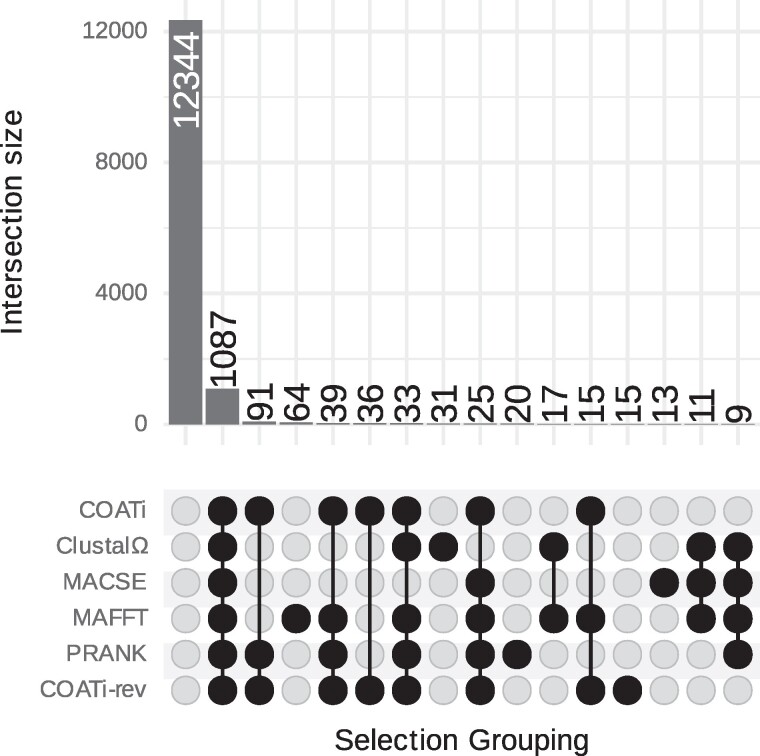
Sequence alignment is an essential method in bioinformatics and the basis of many analyses, including phylogenetic inference, ancestral sequence reconstruction, and gene annotation. Sequencing artifacts and errors made during genome assembly, such as abiological frameshifts and incorrect early stop codons, can impact downstream analyses leading to erroneous conclusions in comparative and functional genomic studies. More significantly, while indels can occur both within and between codons in natural sequences, most amino-acid- and codon-based aligners assume that indels only occur between codons. This mismatch between biology and alignment algorithms produces suboptimal alignments and errors in downstream analyses. To address these issues, we present COATi, a statistical, codon-aware pairwise aligner that supports complex insertion-deletion models and can handle artifacts present in genomic data. COATi allows users to reduce the amount of discarded data while generating more accurate sequence alignments. COATi can infer indels both within and between codons, leading to improved sequence alignments. We applied COATi to a dataset containing orthologous protein-coding sequences from humans and gorillas and conclude that 41% of indels occurred between codons, agreeing with previous work in other species. We also applied COATi to semiempirical benchmark alignments and find that it outperforms several popular alignment programs on several measures of alignment quality and accuracy.
序列比对是生物信息学中的一种基本方法,也是许多分析的基础,包括系统发育推断、祖先序列重建和基因注释。在基因组组装过程中产生的测序伪影和错误,如非生物框架移位和不正确的早期终止密码子,会影响下游分析,导致比较和功能基因组研究中的错误结论。更重要的是,虽然在自然序列中插入缺失可以发生在密码子内和密码子之间,但大多数基于氨基酸和密码子的比对器假设插入缺失只发生在密码子之间。这种生物学和比对算法之间的不匹配会产生次优的比对和下游分析中的错误。为了解决这些问题,我们提出了 COATi,一种统计的、基于密码子的两两比对器,它支持复杂的插入缺失模型,并可以处理基因组数据中存在的伪影。COATi 允许用户在生成更准确的序列比对的同时减少丢弃的数据量。COATi 可以推断密码子内和密码子之间的插入缺失,从而导致更好的序列比对。我们将 COATi 应用于包含人类和大猩猩同源蛋白编码序列的数据集,并得出结论,41%的插入缺失发生在密码子之间,这与其他物种的先前工作一致。我们还将 COATi 应用于半经验基准比对,并发现它在几个比对质量和准确性的度量上优于几个流行的比对程序。