Miller Justin J, Mallimadugula Upasana L, Zimmerman Maxwell I, Stuchell-Brereton Melissa D, Soranno Andrea, Bowman Gregory R
Departments of Biochemistry & Biophysics and Bioengineering, University of Pennsylvania, Philadelphia, PA 19104, United States.
Department of Biochemistry and Molecular Biophysics, Washington University School of Medicine, St. Louis, Missouri 63110, United States.
bioRxiv. 2024 Jun 3:2024.06.03.597137. doi: 10.1101/2024.06.03.597137.
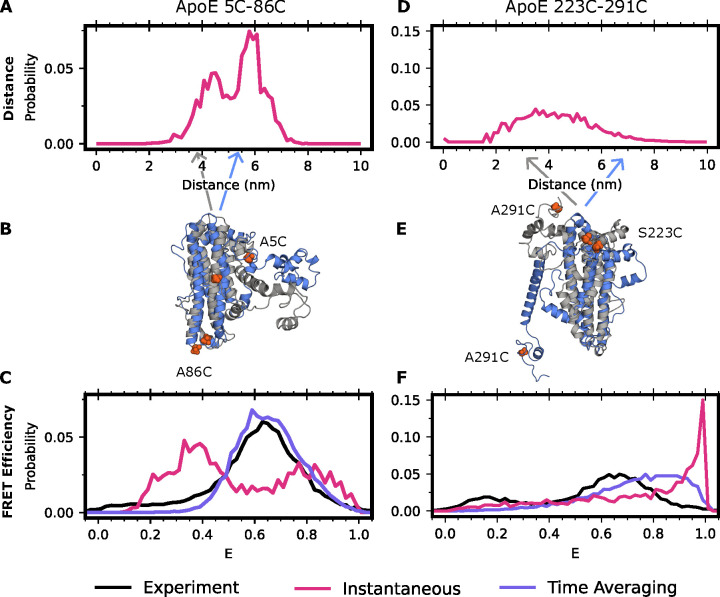
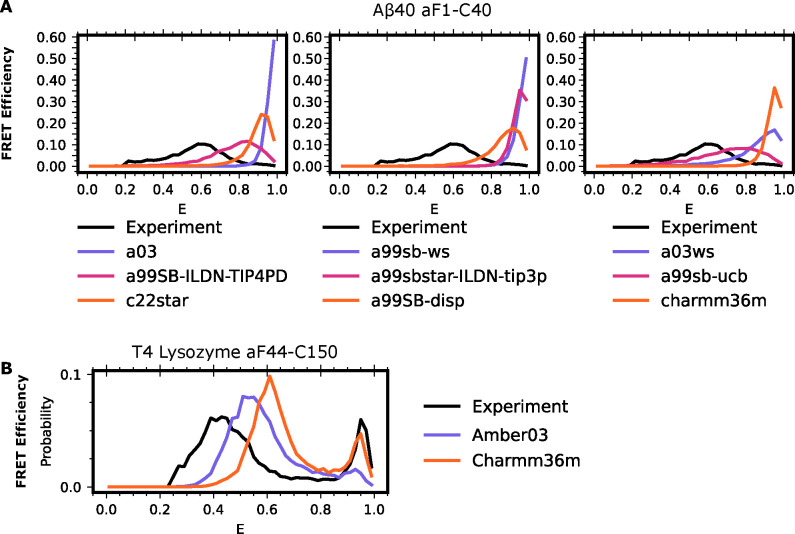
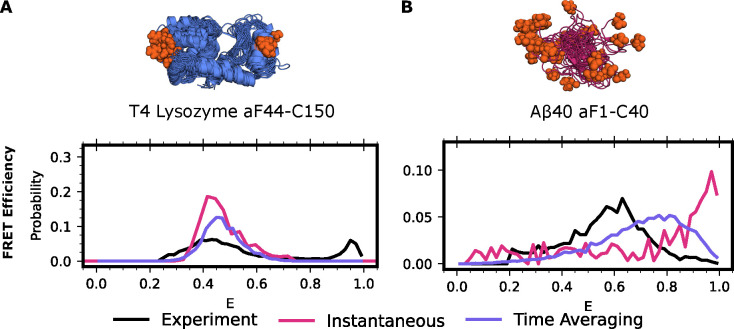
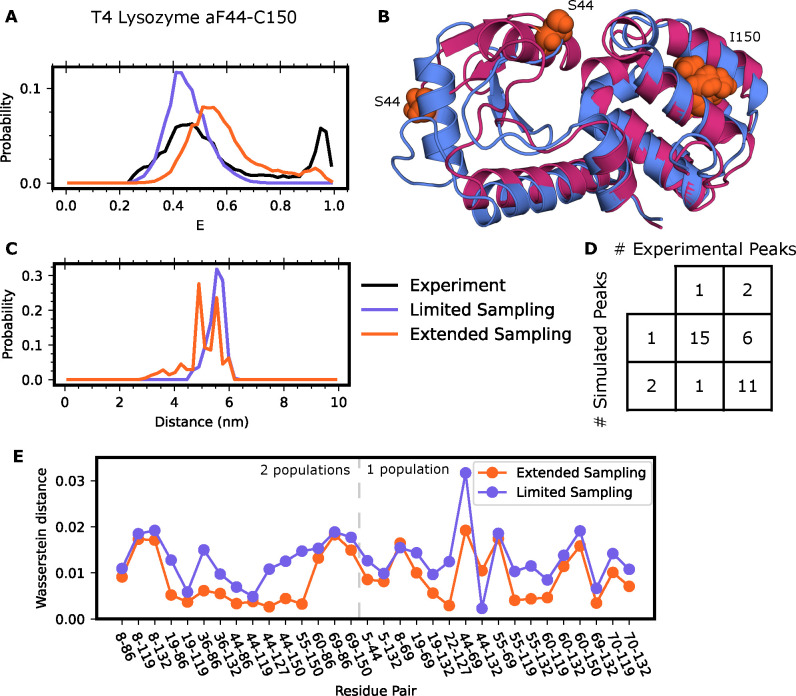
Proteins are dynamic systems whose structural preferences determine their function. Unfortunately, building atomically detailed models of protein structural ensembles remains challenging, limiting our understanding of the relationships between sequence, structure, and function. Combining single molecule Förster resonance energy transfer (smFRET) experiments with molecular dynamics simulations could provide experimentally grounded, all-atom models of a protein's structural ensemble. However, agreement between the two techniques is often insufficient to achieve this goal. Here, we explore whether accounting for important experimental details like averaging across structures sampled during a given smFRET measurement is responsible for this apparent discrepancy. We present an approach to account for this time-averaging by leveraging the kinetic information available from Markov state models of a protein's dynamics. This allows us to accurately assess which timescales are averaged during an experiment. We find this approach significantly improves agreement between simulations and experiments in proteins with varying degrees of dynamics, including the well-ordered protein T4 lysozyme, the partially disordered protein apolipoprotein E (ApoE), and a disordered amyloid protein (Aβ40). We find evidence for hidden states that are not apparent in smFRET experiments because of time averaging with other structures, akin to states in fast exchange in NMR, and evaluate different force fields. Finally, we show how remaining discrepancies between computations and experiments can be used to guide additional simulations and build structural models for states that were previously unaccounted for. We expect our approach will enable combining simulations and experiments to understand the link between sequence, structure, and function in many settings.
蛋白质是动态系统,其结构偏好决定了它们的功能。不幸的是,构建蛋白质结构集合的原子级详细模型仍然具有挑战性,这限制了我们对序列、结构和功能之间关系的理解。将单分子荧光共振能量转移(smFRET)实验与分子动力学模拟相结合,可以提供基于实验的蛋白质结构集合的全原子模型。然而,这两种技术之间的一致性往往不足以实现这一目标。在这里,我们探讨考虑重要的实验细节,如在给定的smFRET测量过程中对采样结构进行平均,是否是造成这种明显差异的原因。我们提出了一种方法,通过利用蛋白质动力学的马尔可夫状态模型中可用的动力学信息来考虑这种时间平均。这使我们能够准确评估实验过程中哪些时间尺度被平均了。我们发现这种方法显著提高了具有不同程度动力学的蛋白质的模拟和实验之间的一致性,包括结构有序的蛋白质T4溶菌酶、部分无序的蛋白质载脂蛋白E(ApoE)和无序的淀粉样蛋白(Aβ40)。我们发现了由于与其他结构的时间平均而在smFRET实验中不明显的隐藏状态的证据,类似于NMR中快速交换的状态,并评估了不同的力场。最后,我们展示了如何利用计算和实验之间的剩余差异来指导额外的模拟,并为以前未考虑的状态构建结构模型。我们期望我们的方法将能够在许多情况下结合模拟和实验来理解序列、结构和功能之间的联系。