Departments of Biochemistry & Biophysics and Bioengineering, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States.
Department of Biochemistry and Molecular Biophysics, Washington University in St. Louis, St. Louis, Missouri 63130, United States.
J Chem Theory Comput. 2024 Feb 13;20(3):1036-1050. doi: 10.1021/acs.jctc.3c00870. Epub 2024 Jan 31.
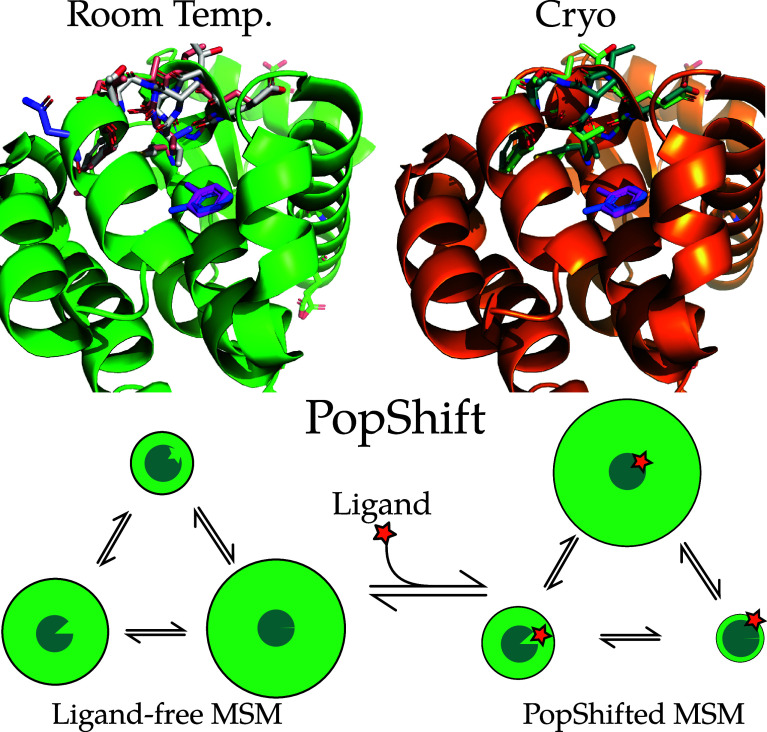
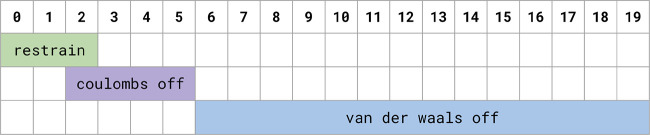
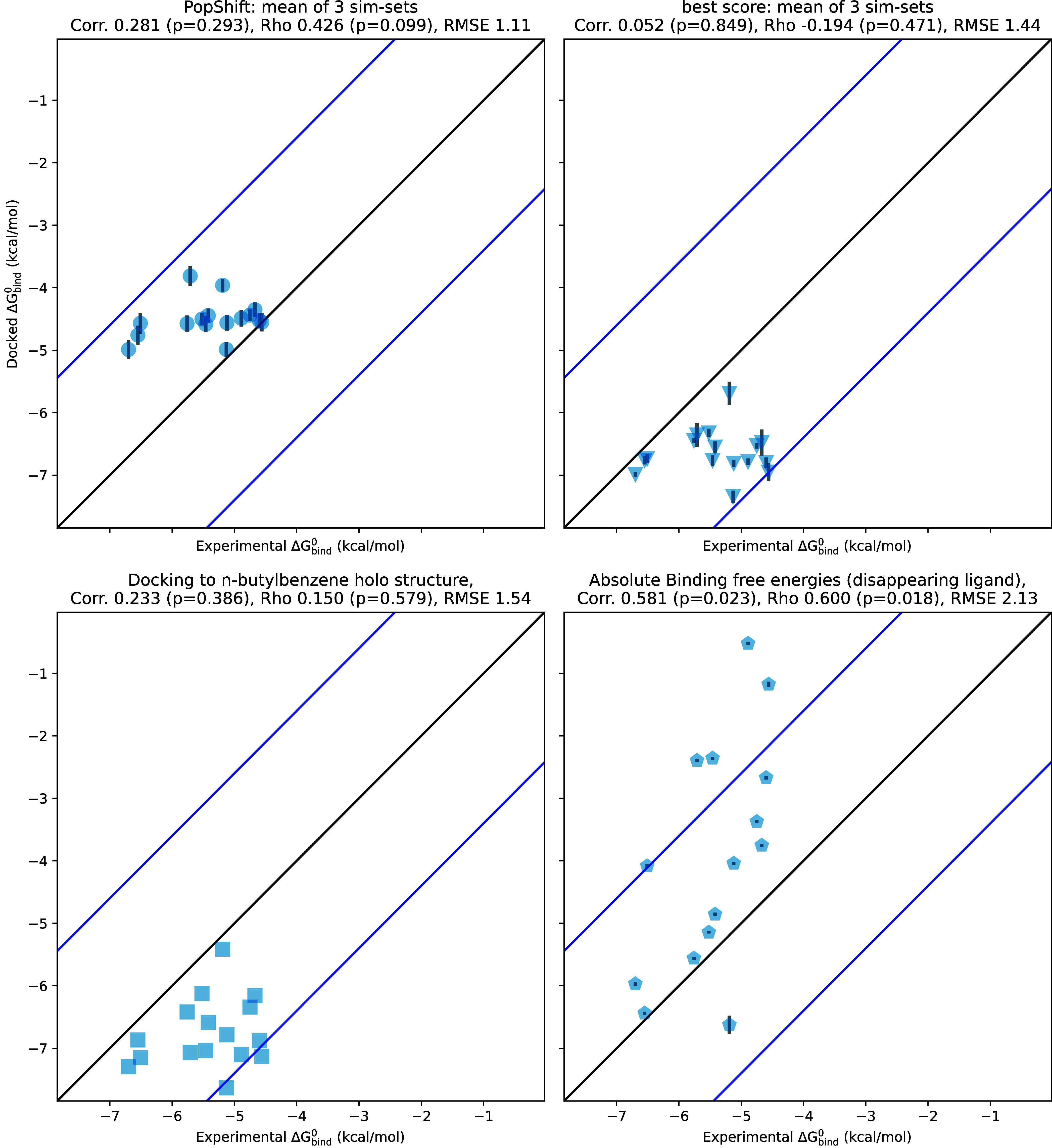
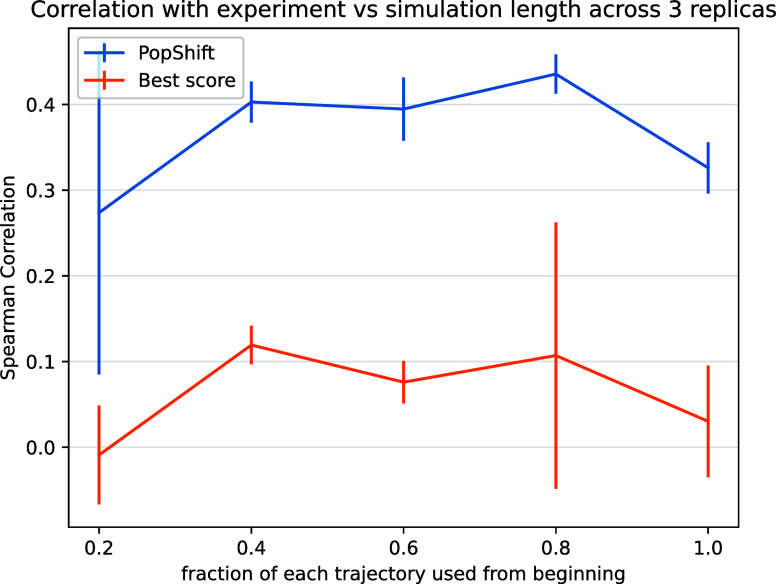
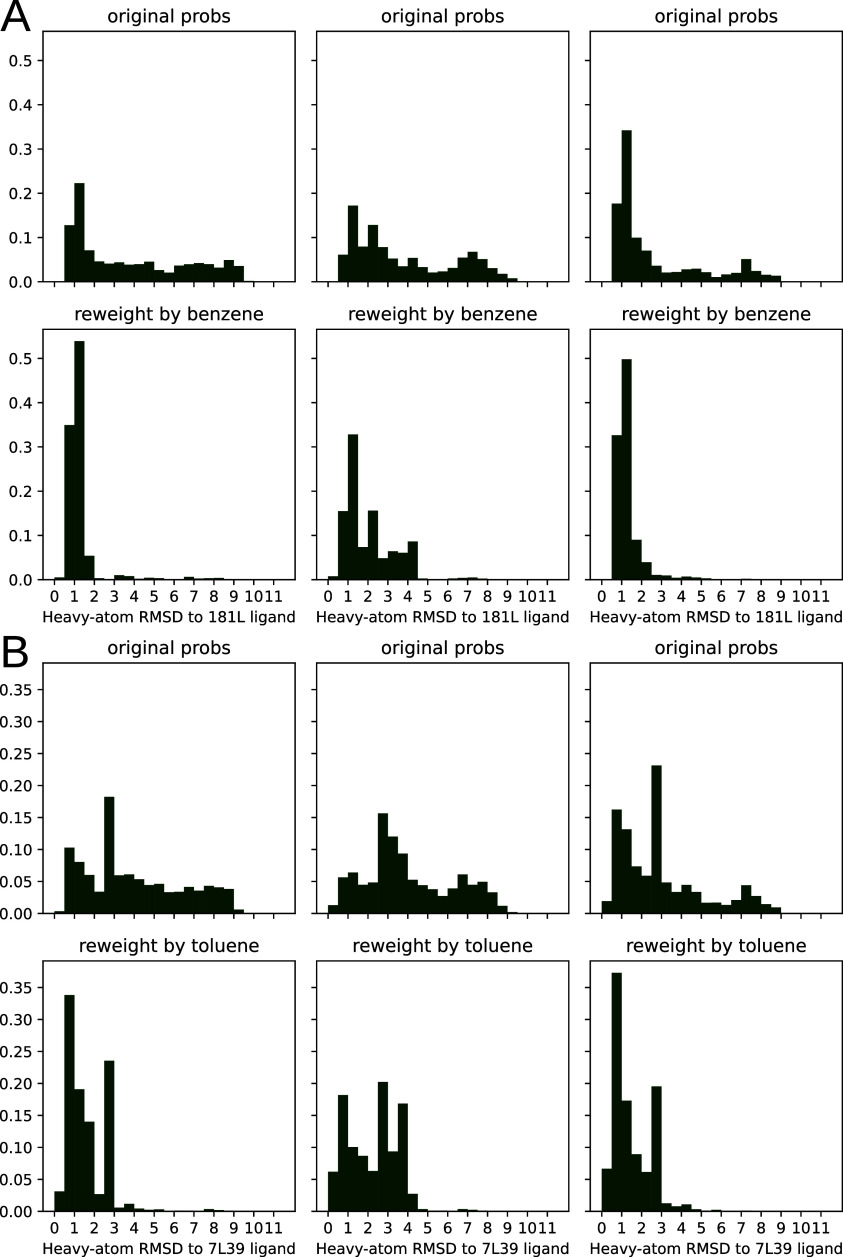
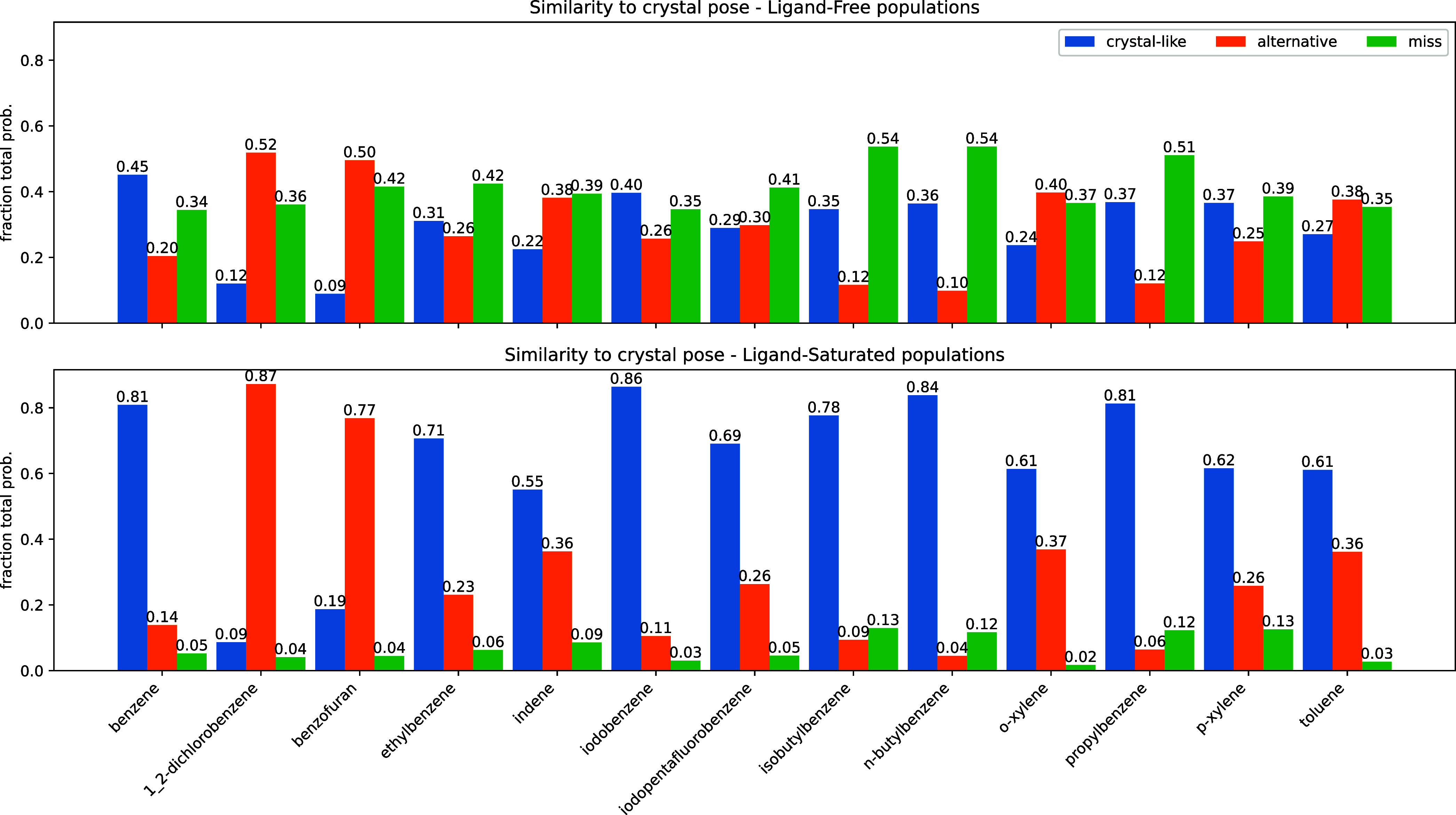
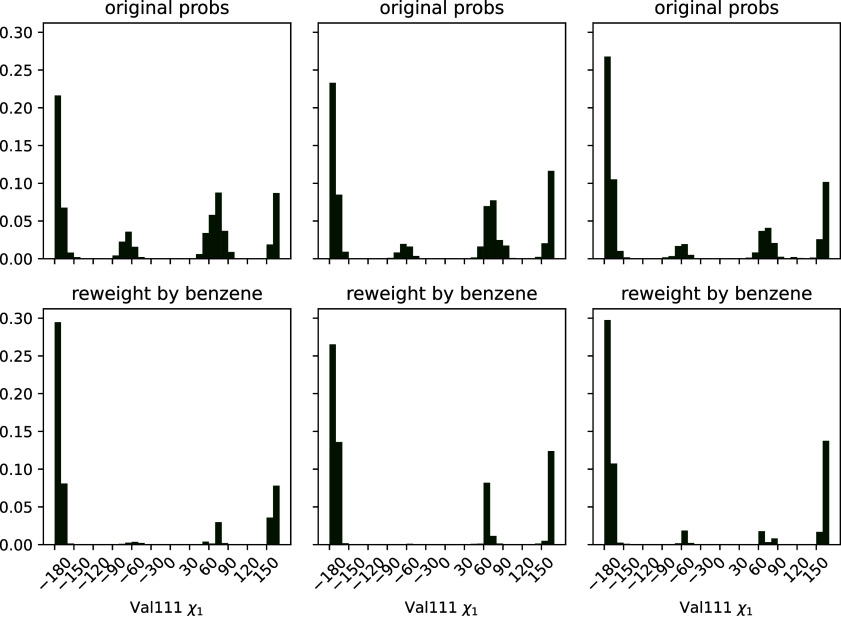
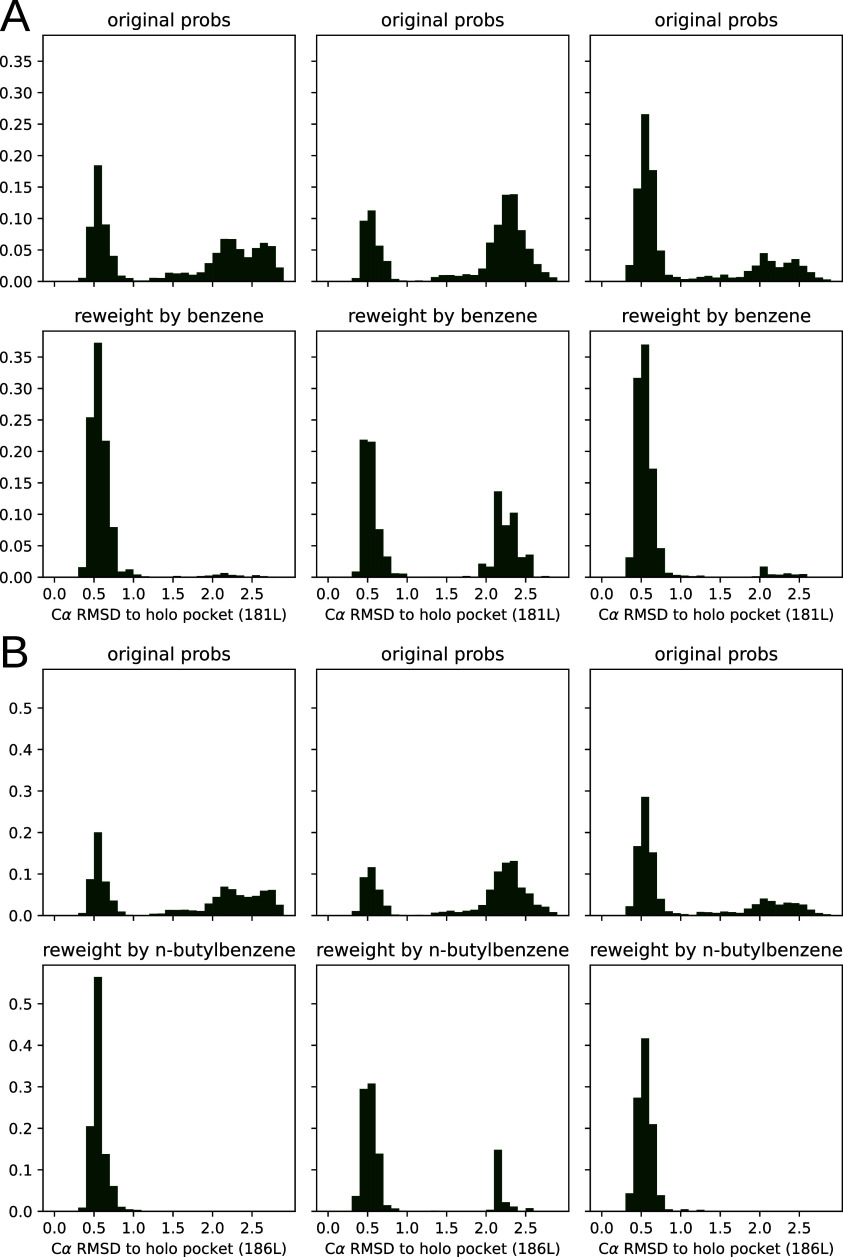
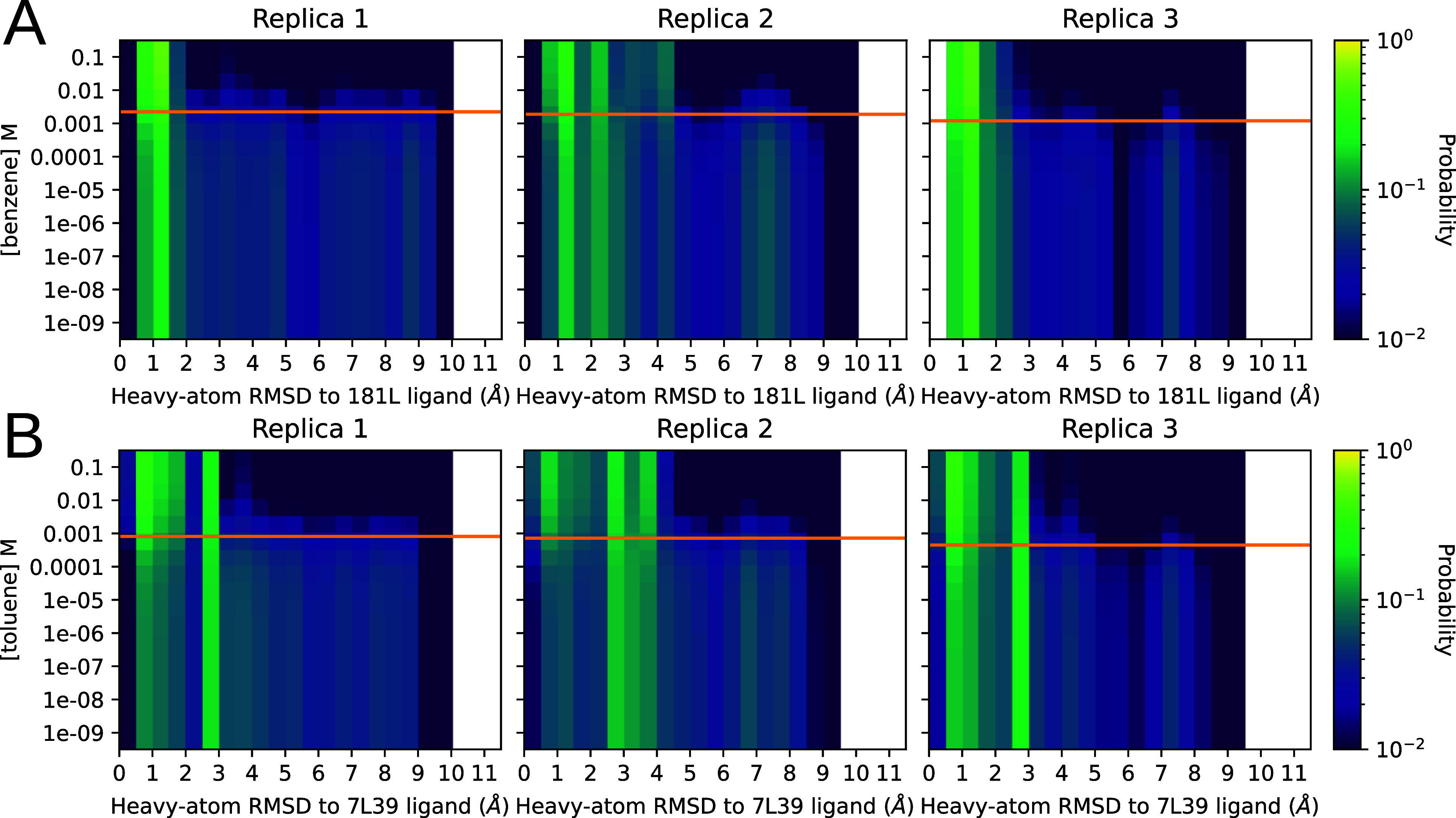
Obtaining accurate binding free energies from screens has been a long-standing goal for the computational chemistry community. However, accuracy and computational cost are at odds with one another, limiting the utility of methods that perform this type of calculation. Many methods achieve massive scale by explicitly or implicitly assuming that the target protein adopts a single structure, or undergoes limited fluctuations around that structure, to minimize computational cost. Others simulate each protein-ligand complex of interest, accepting lower throughput in exchange for better predictions of binding affinities. Here, we present the PopShift framework for accounting for the ensemble of structures a protein adopts and their relative probabilities. Protein degrees of freedom are enumerated once, and then arbitrarily many molecules can be screened against this ensemble. Specifically, we use Markov state models (MSMs) as a compressed representation of a protein's thermodynamic ensemble. We start with a ligand-free MSM and then calculate how addition of a ligand shifts the populations of each protein conformational state based on the strength of the interaction between that protein conformation and the ligand. In this work we use docking to estimate the affinity between a given protein structure and ligand, but any estimator of binding affinities could be used in the PopShift framework. We test PopShift on the classic benchmark pocket T4 Lysozyme L99A. We find that PopShift is more accurate than common strategies, such as docking to a single structure and traditional ensemble docking─producing results that compare favorably with alchemical binding free energy calculations in terms of RMSE but not correlation─and may have a more favorable computational cost profile in some applications. In addition to predicting binding free energies and ligand poses, PopShift also provides insight into how the probability of different protein structures is shifted upon addition of various concentrations of ligand, providing a platform for predicting affinities and allosteric effects of ligand binding. Therefore, we expect PopShift will be valuable for hit finding and for providing insight into phenomena like allostery.
从筛选中获得准确的结合自由能一直是计算化学界的长期目标。然而,准确性和计算成本相互矛盾,限制了执行此类计算的方法的实用性。许多方法通过显式或隐式地假设目标蛋白采用单一结构或围绕该结构进行有限的波动来实现大规模,以最小化计算成本。其他方法模拟每个感兴趣的蛋白质-配体复合物,以牺牲结合亲和力的更好预测为代价接受较低的通量。在这里,我们提出了 PopShift 框架,用于说明蛋白质采用的结构集合及其相对概率。蛋白质自由度仅枚举一次,然后可以针对该集合对任意数量的分子进行筛选。具体来说,我们使用马尔可夫状态模型 (MSM) 作为蛋白质热力学集合的压缩表示。我们从无配体的 MSM 开始,然后根据该蛋白质构象与配体之间相互作用的强度计算添加配体如何改变每个蛋白质构象状态的种群。在这项工作中,我们使用对接来估计给定蛋白质结构与配体之间的亲和力,但任何结合亲和力的估计器都可以在 PopShift 框架中使用。我们在经典的口袋 T4 溶菌酶 L99A 基准上测试了 PopShift。我们发现 PopShift 比常见策略更准确,例如对接单一结构和传统的整体对接─产生的结果在 RMSE 方面与化学结合自由能计算相比具有优势,但在相关性方面没有优势─并且在某些应用中可能具有更有利的计算成本特征。除了预测结合自由能和配体构象外,PopShift 还提供了有关在添加各种浓度的配体时不同蛋白质结构的概率如何变化的见解,为预测配体结合的亲和力和变构效应提供了一个平台。因此,我们预计 PopShift 将对发现命中和深入了解变构等现象具有重要价值。