Fu Xingke, Geng Zhi, Jiao Zhichao, Ding Wei
Beijing National Laboratory for Condensed Matter Physics, Institute of Physics, Chinese Academy of Sciences, Beijing 100190, People's Republic of China.
Beijing Synchrotron Radiation Facility, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, People's Republic of China.
IUCrJ. 2024 Jul 1;11(Pt 4):587-601. doi: 10.1107/S2052252524004846.
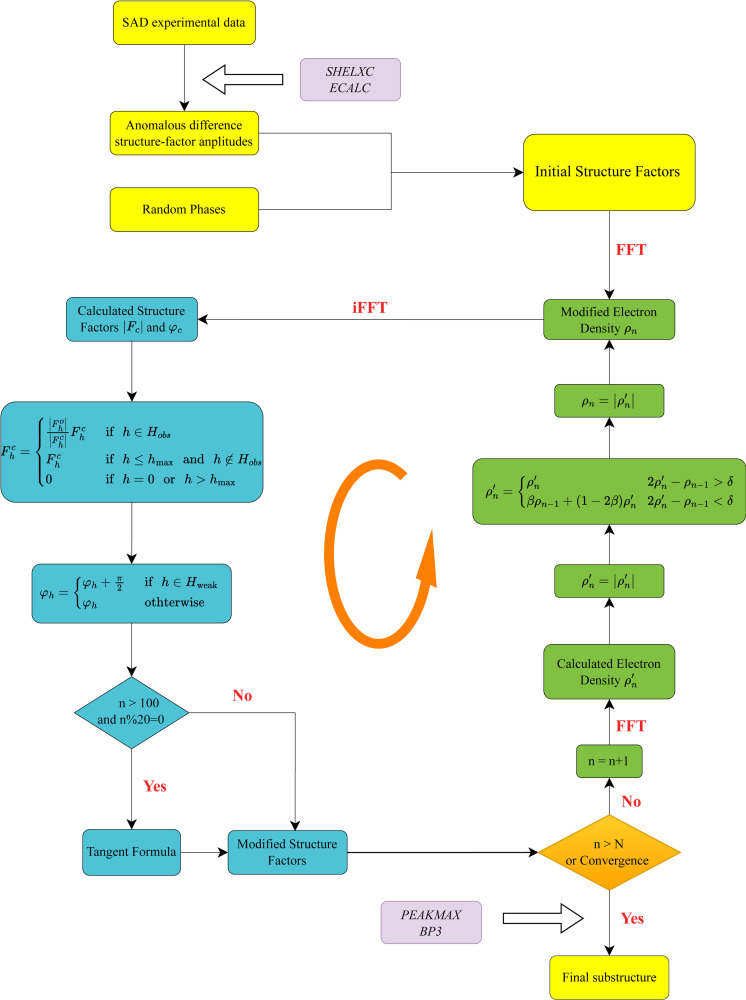
The success of experimental phasing in macromolecular crystallography relies primarily on the accurate locations of heavy atoms bound to the target crystal. To improve the process of substructure determination, a modified phase-retrieval algorithm built on the framework of the relaxed alternating averaged reflection (RAAR) algorithm has been developed. Importantly, the proposed algorithm features a combination of the π-half phase perturbation for weak reflections and enforces the direct-method-based tangent formula for strong reflections in reciprocal space. The proposed algorithm is extensively demonstrated on a total of 100 single-wavelength anomalous diffraction (SAD) experimental datasets, comprising both protein and nucleic acid structures of different qualities. Compared with the standard RAAR algorithm, the modified phase-retrieval algorithm exhibits significantly improved effectiveness and accuracy in SAD substructure determination, highlighting the importance of additional constraints for algorithmic performance. Furthermore, the proposed algorithm can be performed without human intervention under most conditions owing to the self-adaptive property of the input parameters, thus making it convenient to be integrated into the structural determination pipeline. In conjunction with the IPCAS software suite, we demonstrated experimentally that automatic de novo structure determination is possible on the basis of our proposed algorithm.
大分子晶体学中实验相位确定的成功主要依赖于与目标晶体结合的重原子的准确位置。为了改进亚结构确定过程,基于松弛交替平均反射(RAAR)算法框架开发了一种改进的相位恢复算法。重要的是,该算法的特点是对弱反射采用π/2相位扰动,并在倒易空间中对强反射强制执行基于直接法的切线公式。该算法在总共100个单波长反常衍射(SAD)实验数据集上进行了广泛验证,这些数据集包括不同质量的蛋白质和核酸结构。与标准RAAR算法相比,改进后的相位恢复算法在SAD亚结构确定中表现出显著提高的有效性和准确性,突出了附加约束对算法性能的重要性。此外,由于输入参数的自适应特性,该算法在大多数情况下无需人工干预即可执行,因此便于集成到结构确定流程中。结合IPCAS软件套件,我们通过实验证明,基于我们提出的算法可以实现自动从头结构确定。