State Key Laboratory of Genetic Engineering, Human Phenome Institute, Zhangjiang Fudan International Innovation Center, School of Life Science, Fudan University, Shanghai, 200438, China.
Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX, USA.
Mol Genet Genomics. 2024 Jul 7;299(1):65. doi: 10.1007/s00438-024-02158-x.
A large number of challenging medically relevant genes (CMRGs) are situated in complex or highly repetitive regions of the human genome, hindering comprehensive characterization of genetic variants using next-generation sequencing technologies. In this study, we employed long-read sequencing technology, extensively utilized in studying complex genomic regions, to characterize genetic alterations, including short variants (single nucleotide variants and short insertions and deletions) and copy number variations, in 370 CMRGs across 41 individuals from 19 global populations.
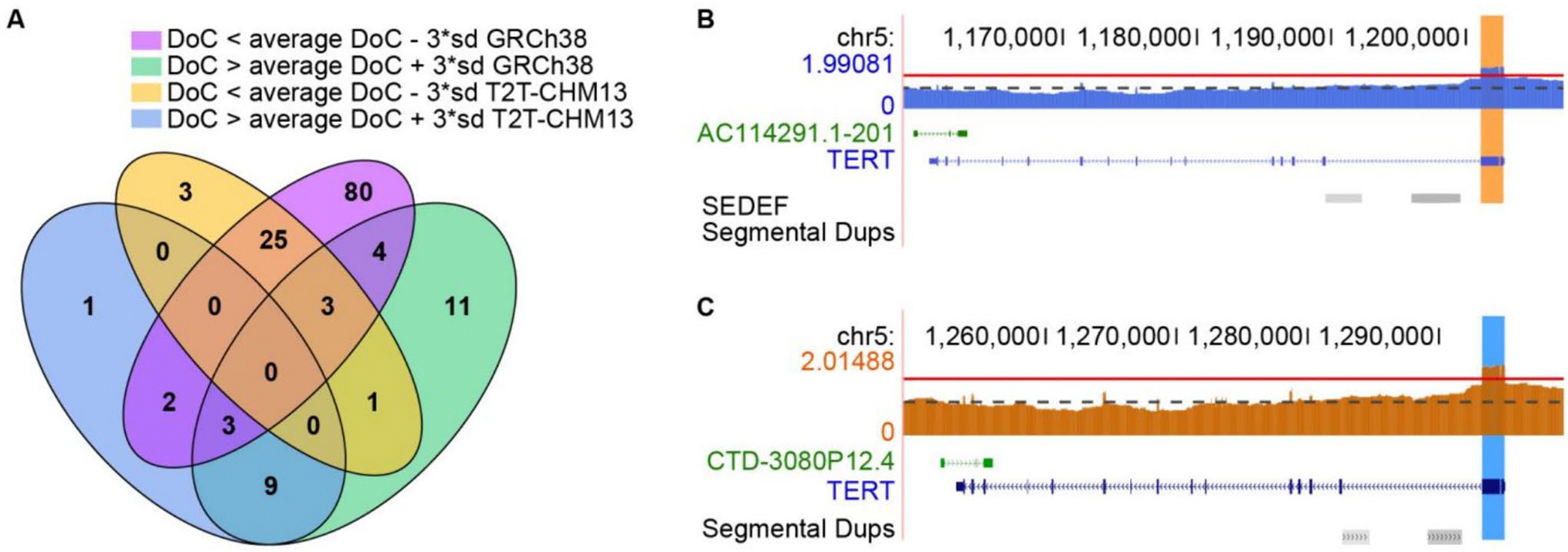
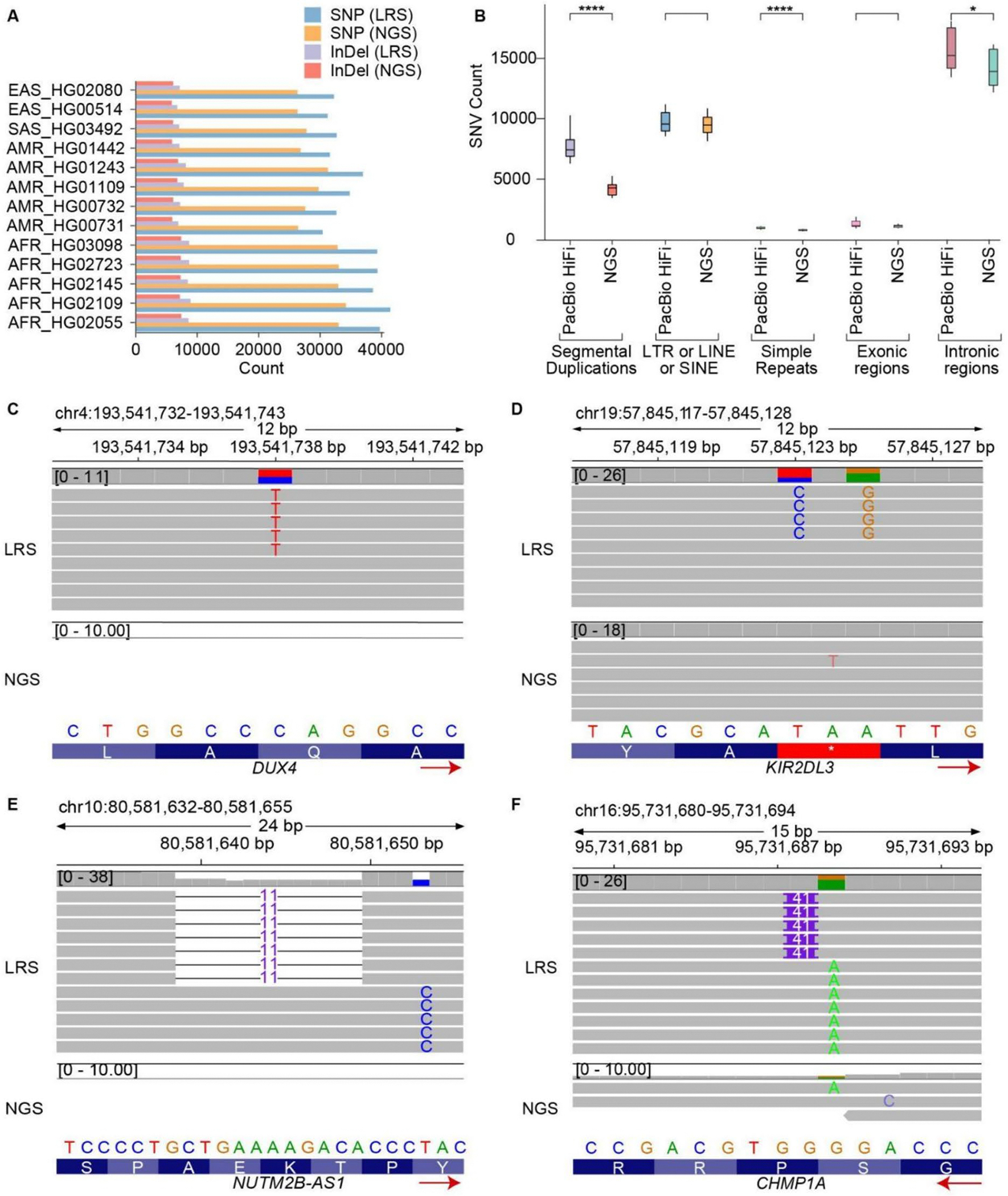
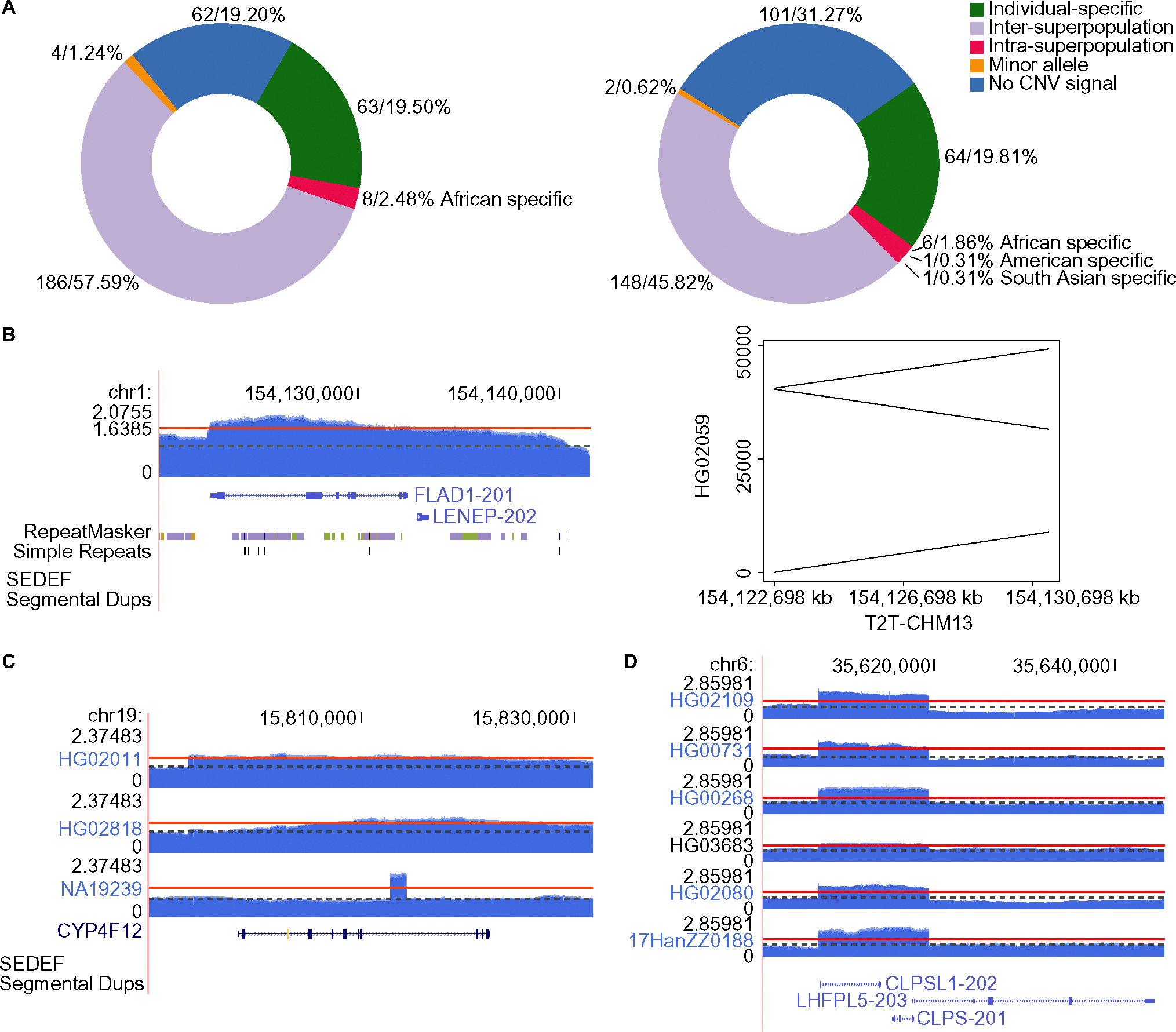
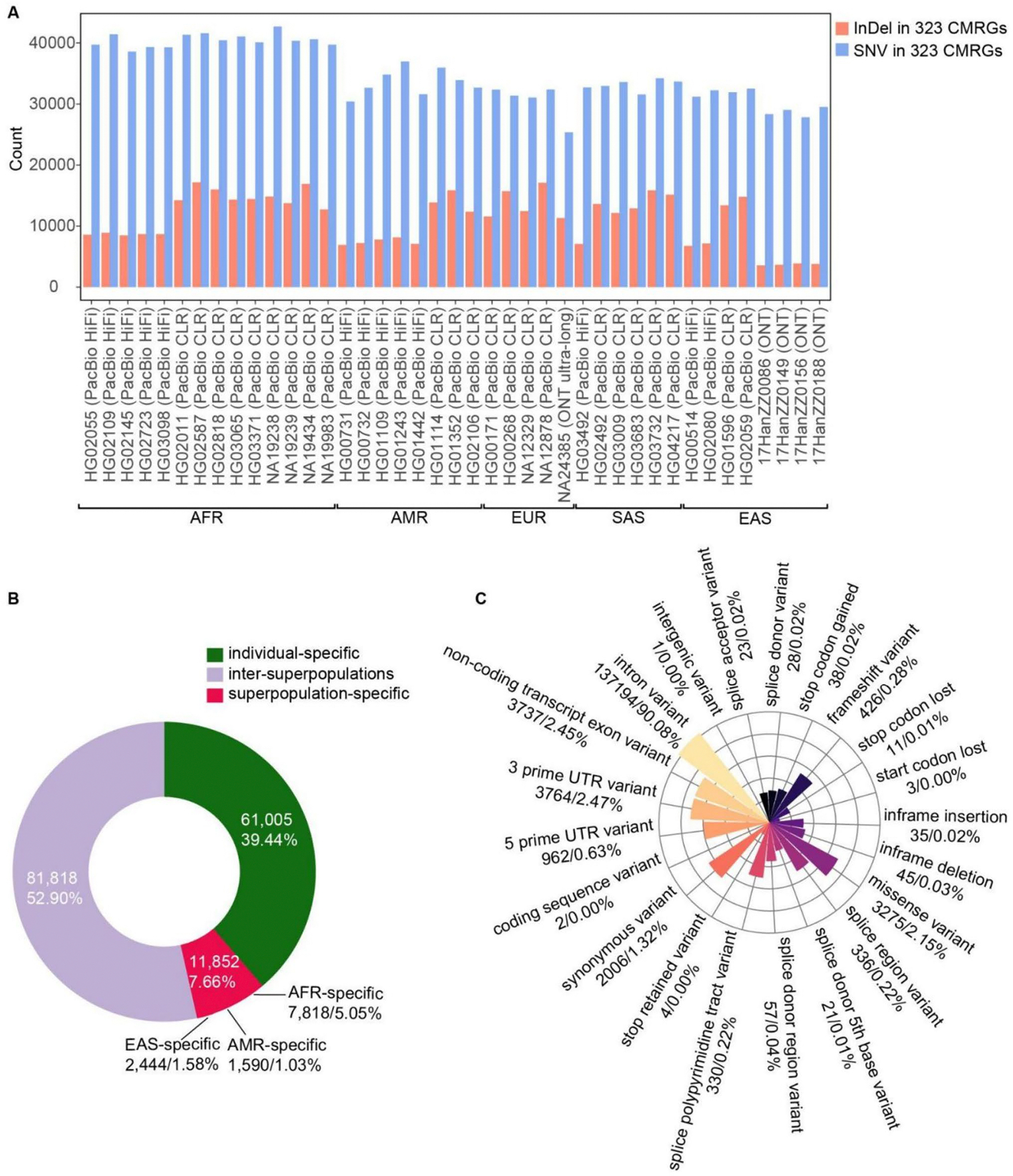
Our analysis revealed high levels of genetic variants in CMRGs, with 68.73% exhibiting copy number variations and 65.20% containing short variants that may disrupt protein function across individuals. Such variants can influence pharmacogenomics, genetic disease susceptibility, and other clinical outcomes. We observed significant differences in CMRG variation across populations, with individuals of African ancestry harboring the highest number of copy number variants and short variants compared to samples from other continents. Notably, 15.79% to 33.96% of short variants were exclusively detectable through long-read sequencing. While the T2T-CHM13 reference genome significantly improved the assembly of CMRG regions, thereby facilitating variant detection in these regions, some regions still lacked resolution.
Our results provide an important reference for future clinical and pharmacogenetic studies, highlighting the need for a comprehensive representation of global genetic diversity in the reference genome and improved variant calling techniques to fully resolve medically relevant genes.
大量具有挑战性的医学相关基因(CMRGs)位于人类基因组的复杂或高度重复区域,这阻碍了使用下一代测序技术对遗传变异进行全面表征。在这项研究中,我们采用了长读测序技术,该技术广泛用于研究复杂基因组区域,以表征 41 名来自 19 个全球人群的个体中 370 个 CMRG 中的遗传改变,包括短变异(单核苷酸变异和短插入和缺失)和拷贝数变异。
我们的分析显示 CMRG 中存在高水平的遗传变异,其中 68.73%表现出拷贝数变异,65.20%含有可能破坏个体蛋白功能的短变异。这些变异会影响药物基因组学、遗传疾病易感性和其他临床结果。我们观察到 CMRG 变异在人群之间存在显著差异,与来自其他大陆的样本相比,非洲裔个体携带的拷贝数变异和短变异数量最多。值得注意的是,15.79%至 33.96%的短变异只能通过长读测序来检测。虽然 T2T-CHM13 参考基因组显著提高了 CMRG 区域的组装,从而有助于在这些区域中检测变异,但某些区域仍缺乏分辨率。
我们的研究结果为未来的临床和药物遗传学研究提供了重要参考,突出了在参考基因组中全面代表全球遗传多样性和改进变异调用技术的必要性,以充分解析医学相关基因。