Department of Biochemistry and Molecular Biology, Sidney Kimmel Medical College, Thomas Jefferson University, Philadelphia, PA 19107.
Proc Natl Acad Sci U S A. 2024 Jul 16;121(29):e2321408121. doi: 10.1073/pnas.2321408121. Epub 2024 Jul 8.
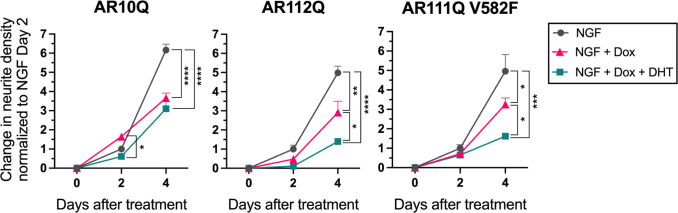
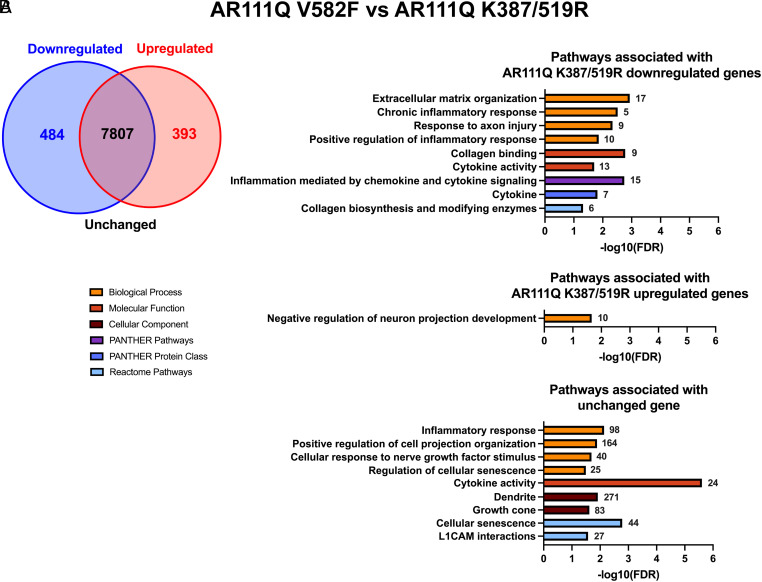
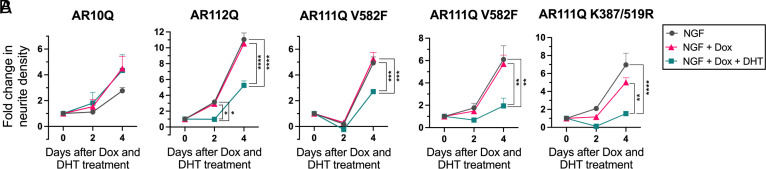
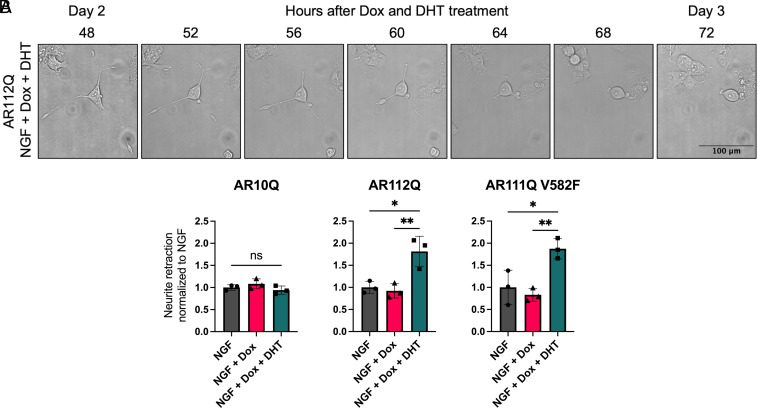
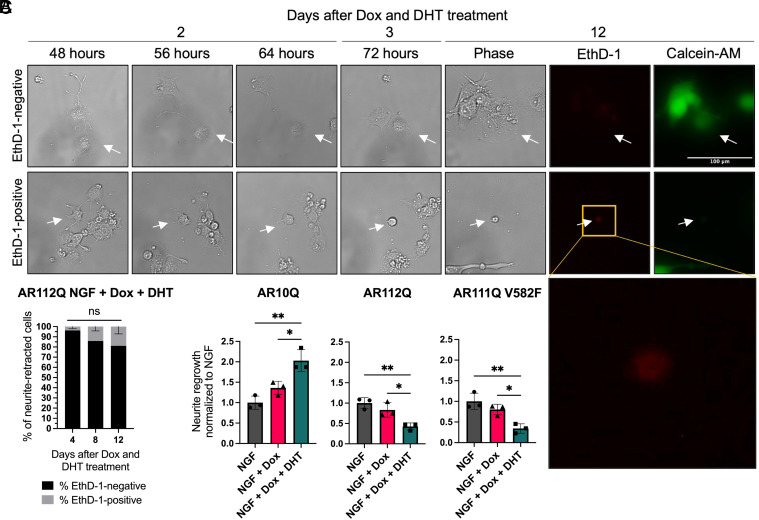
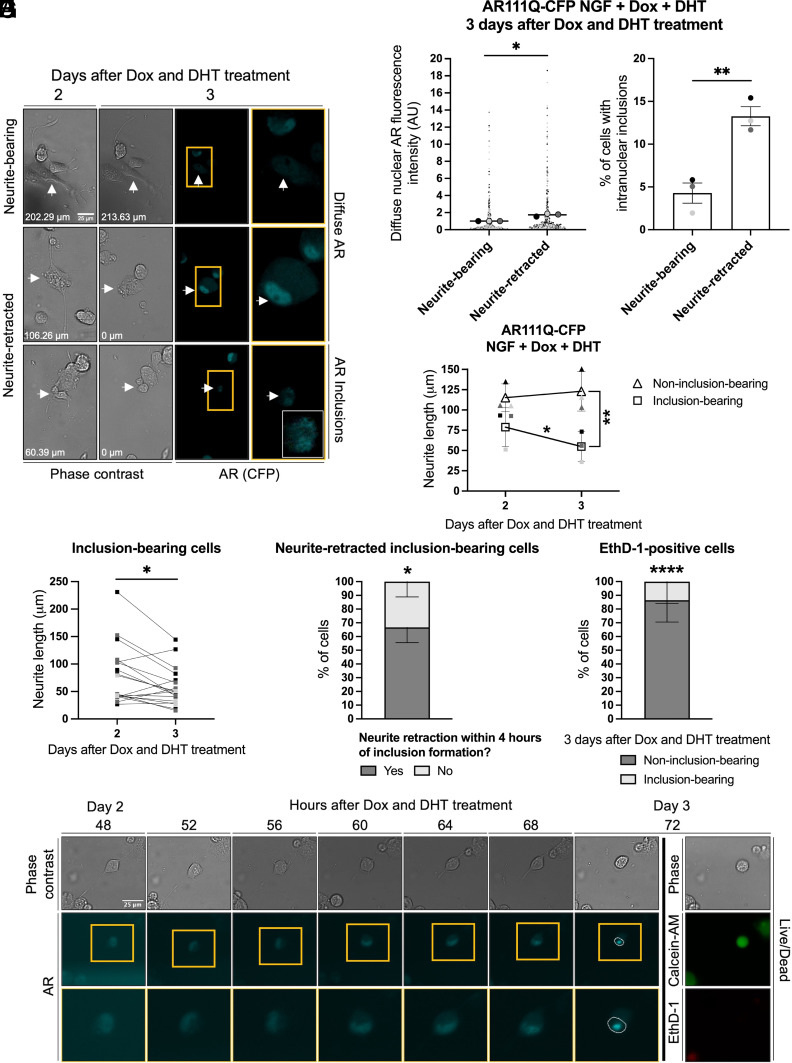
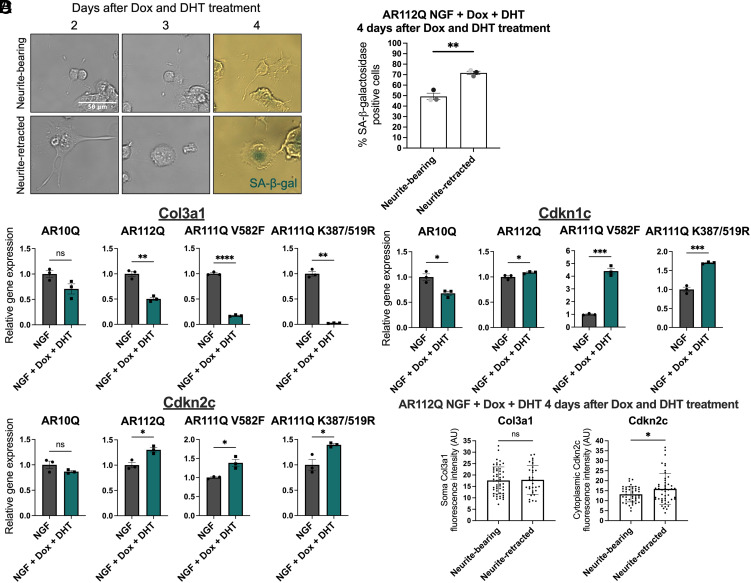
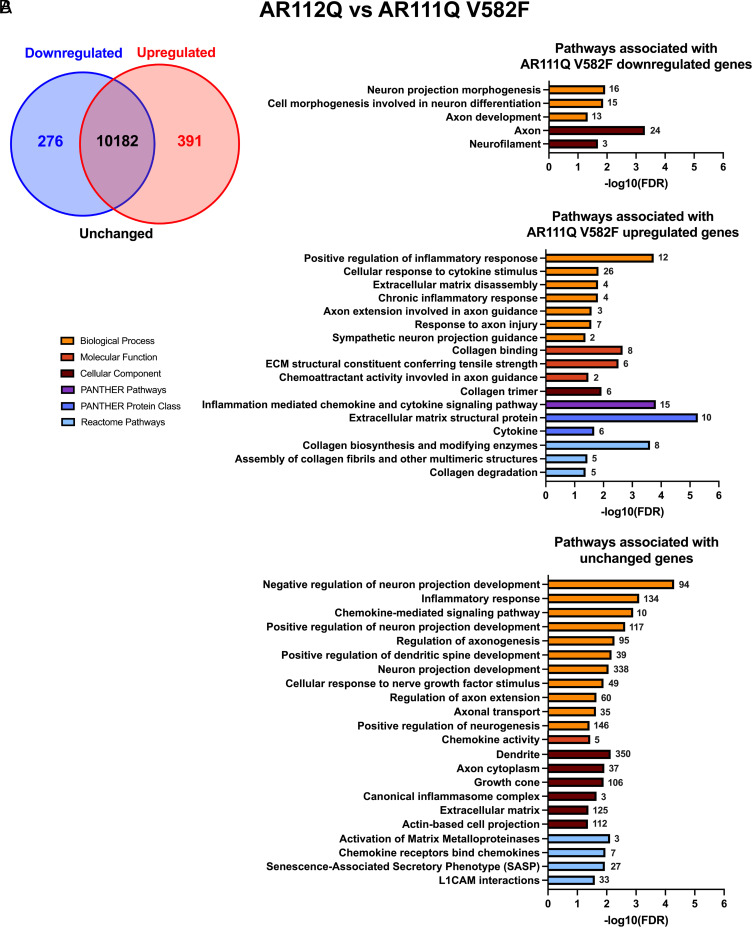
Spinal and bulbar muscular atrophy (SBMA) is a slowly progressing neuromuscular disease caused by a polyglutamine (polyQ)-encoding CAG trinucleotide repeat expansion in the androgen receptor (AR) gene, leading to AR aggregation, lower motor neuron death, and muscle atrophy. AR is a ligand-activated transcription factor that regulates neuronal architecture and promotes axon regeneration; however, whether AR transcriptional functions contribute to disease pathogenesis is not fully understood. Using a differentiated PC12 cell model of SBMA, we identified dysfunction of polyQ-expanded AR in its regulation of neurite growth and maintenance. Specifically, we found that in the presence of androgens, polyQ-expanded AR inhibited neurite outgrowth, induced neurite retraction, and inhibited neurite regrowth. This dysfunction was independent of polyQ-expanded AR transcriptional activity at androgen response elements (ARE). We further showed that the formation of polyQ-expanded AR intranuclear inclusions promoted neurite retraction, which coincided with reduced expression of the neuronal differentiation marker β-III-Tubulin. Finally, we revealed that cell death is not the primary outcome for cells undergoing neurite retraction; rather, these cells become senescent. Our findings reveal that mechanisms independent of AR canonical transcriptional activity underly neurite defects in a cell model of SBMA and identify senescence as a pathway implicated in this pathology. These findings suggest that in the absence of a role for AR canonical transcriptional activity in the SBMA pathologies described here, the development of SBMA therapeutics that preserve this activity may be desirable. This approach may be broadly applicable to other polyglutamine diseases such as Huntington's disease and spinocerebellar ataxias.
脊髓延髓肌肉萎缩症(SBMA)是一种进行性缓慢的神经肌肉疾病,由雄激素受体(AR)基因中编码多聚谷氨酰胺(polyQ)的 CAG 三核苷酸重复扩展引起,导致 AR 聚集、运动神经元死亡和肌肉萎缩。AR 是一种配体激活的转录因子,调节神经元结构并促进轴突再生;然而,AR 转录功能是否有助于疾病发病机制尚不完全清楚。我们使用 SBMA 的分化 PC12 细胞模型,确定了多聚 Q 扩展的 AR 在其调节神经突生长和维持中的功能障碍。具体来说,我们发现,在雄激素存在的情况下,多聚 Q 扩展的 AR 抑制神经突生长,诱导神经突回缩,并抑制神经突再生。这种功能障碍与雄激素反应元件(ARE)处的多聚 Q 扩展 AR 转录活性无关。我们进一步表明,多聚 Q 扩展的 AR 核内包含物的形成促进了神经突回缩,这与神经元分化标志物 β-III-Tubulin 的表达减少相一致。最后,我们揭示了细胞死亡不是经历神经突回缩的细胞的主要结果;相反,这些细胞进入衰老状态。我们的发现表明,在 SBMA 细胞模型中,独立于 AR 经典转录活性的机制导致神经突缺陷,并确定衰老作为涉及这种病理的途径。这些发现表明,在缺乏 AR 经典转录活性在本文描述的 SBMA 病理中的作用的情况下,开发保留这种活性的 SBMA 治疗方法可能是可取的。这种方法可能广泛适用于其他多聚谷氨酰胺疾病,如亨廷顿病和脊髓小脑共济失调。