Manaithiya Ajay, Bhowmik Ratul, Acharjee Satarupa, Sharma Sameer, Kumar Sunil, Imran Mohd, Mathew Bijo, Parkkila Seppo, Aspatwar Ashok
Faculty of Medicine and Health Technology, Tampere University, Tampere, Finland.
Department of Pharmacy, NSHM Knowledge Campus, Kolkata-Group of Institutions, Kolkata, West Bengal 700053, India.
Comput Struct Biotechnol J. 2024 Jul 6;23:2811-2836. doi: 10.1016/j.csbj.2024.07.006. eCollection 2024 Dec.
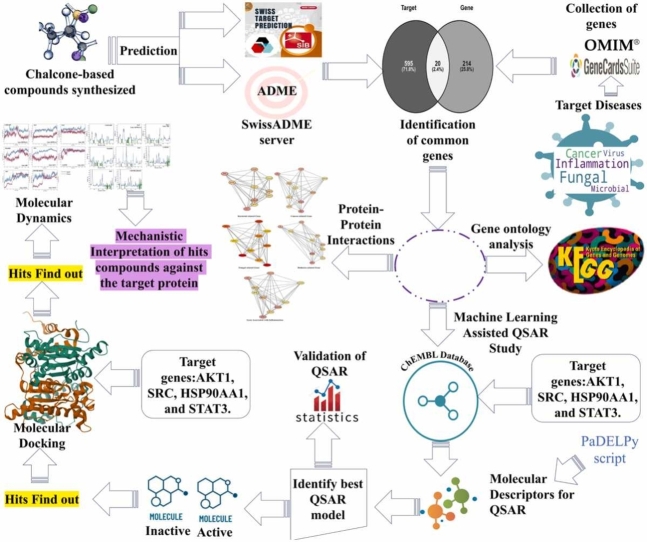
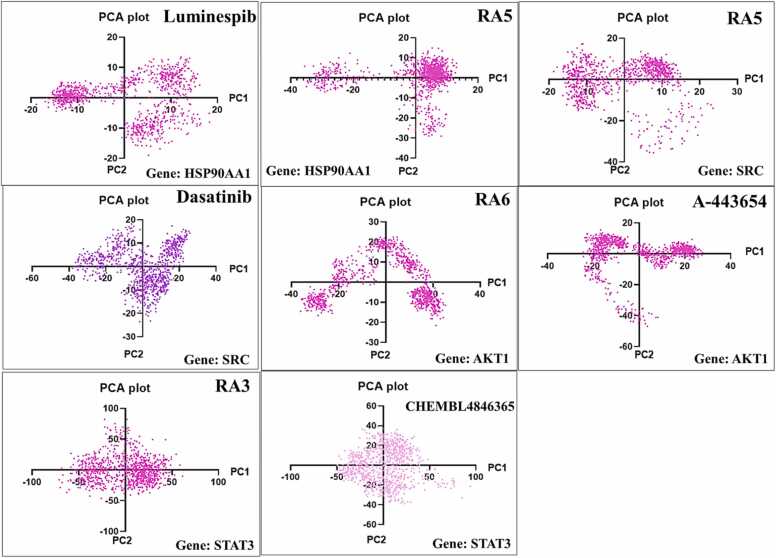
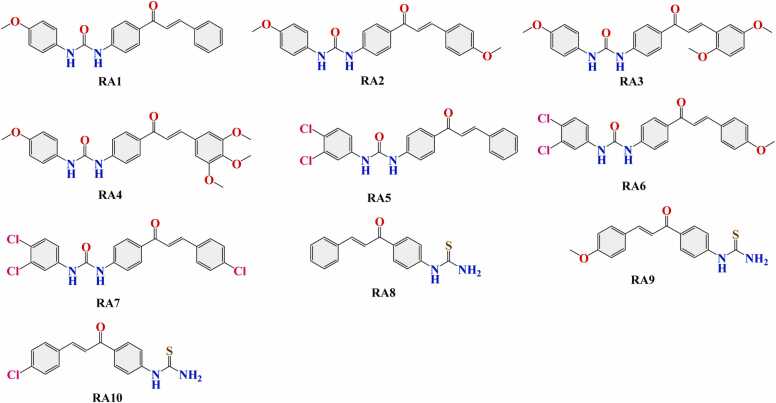
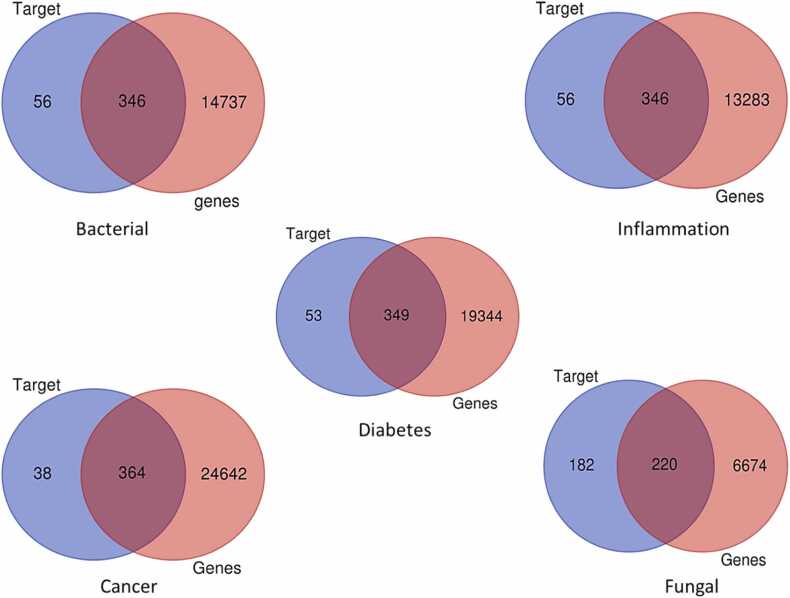
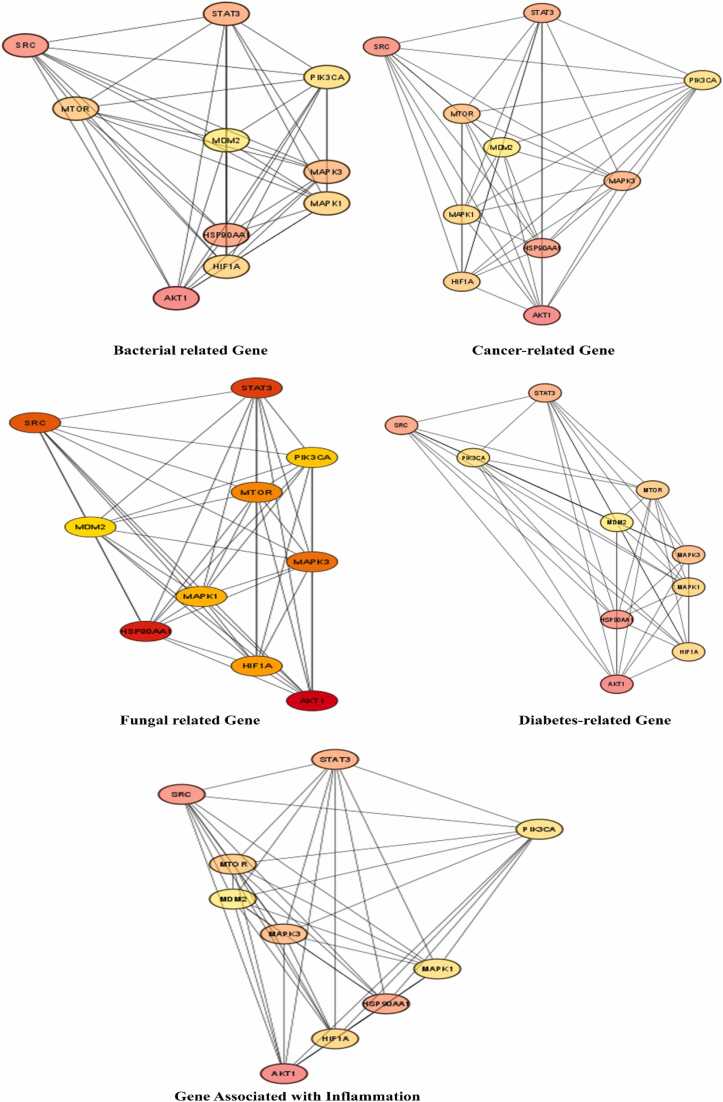
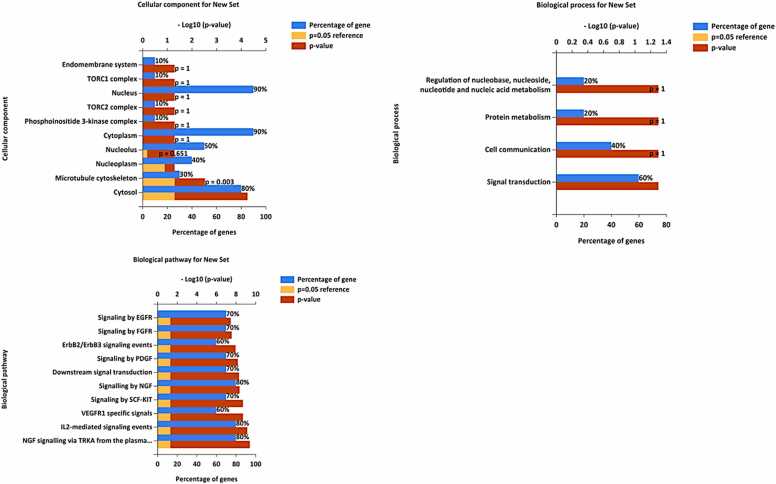
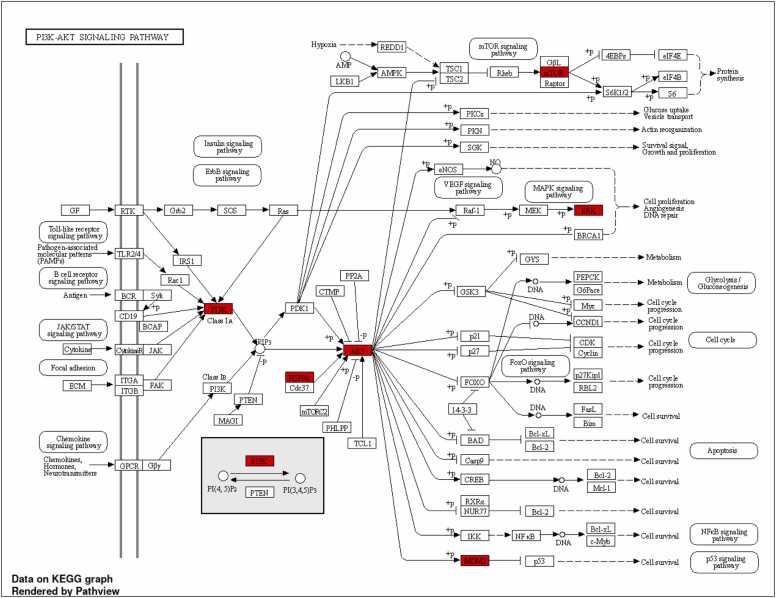
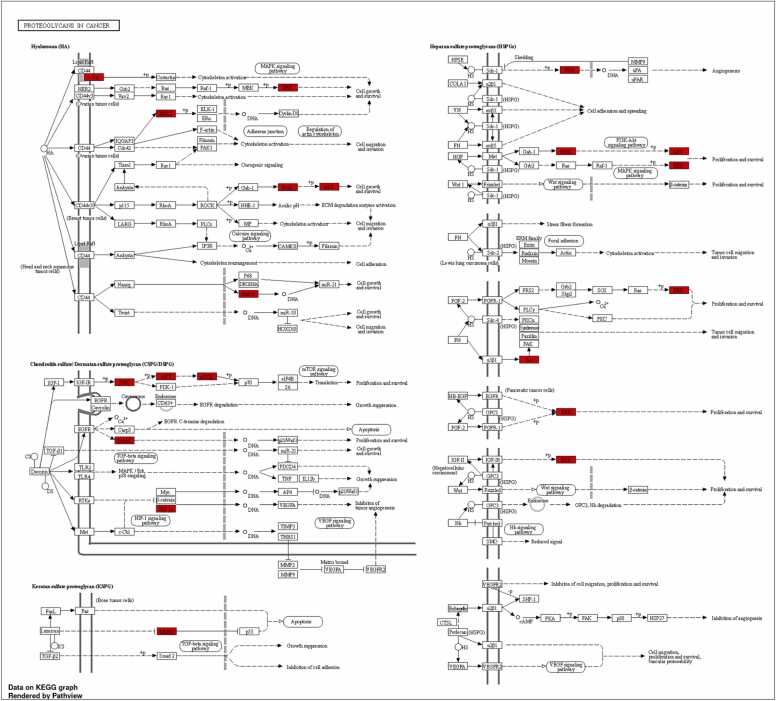
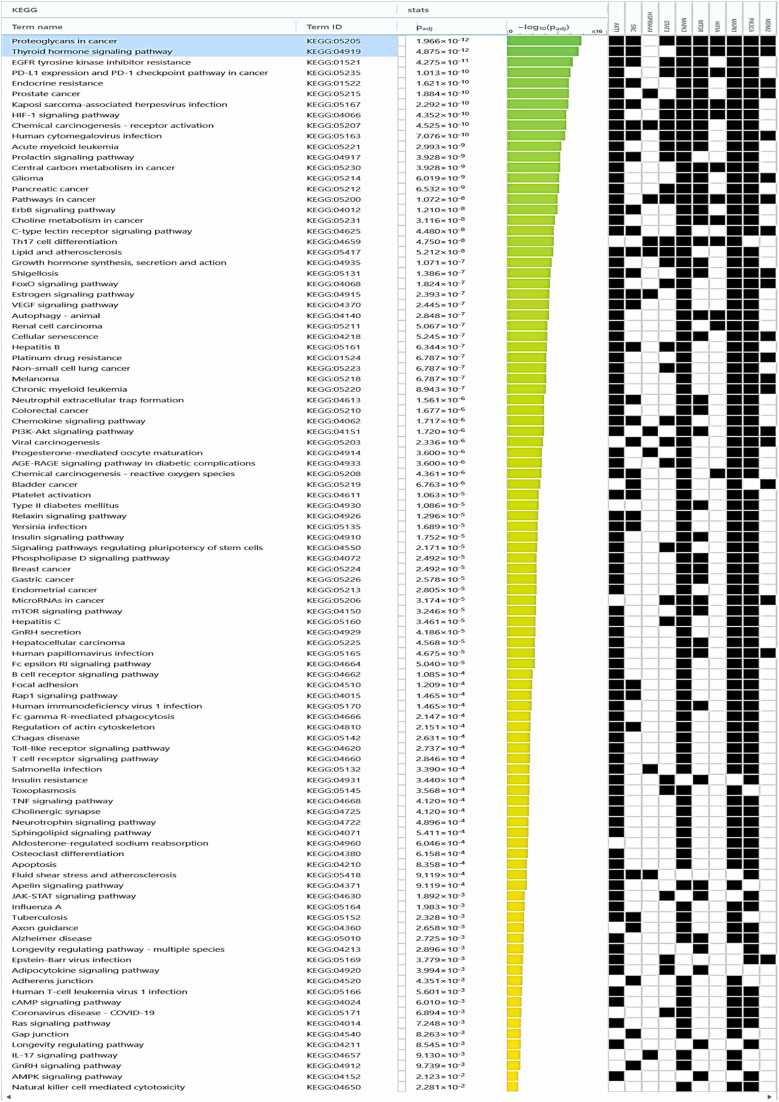
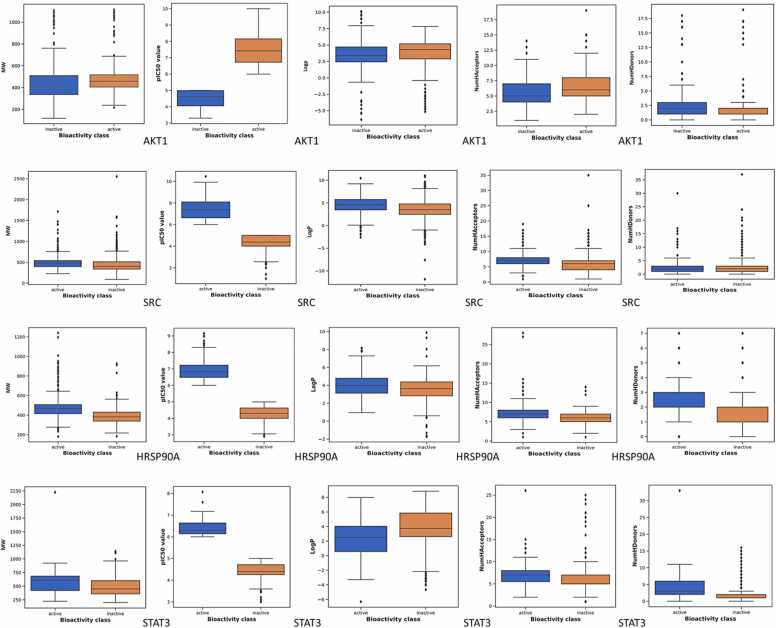
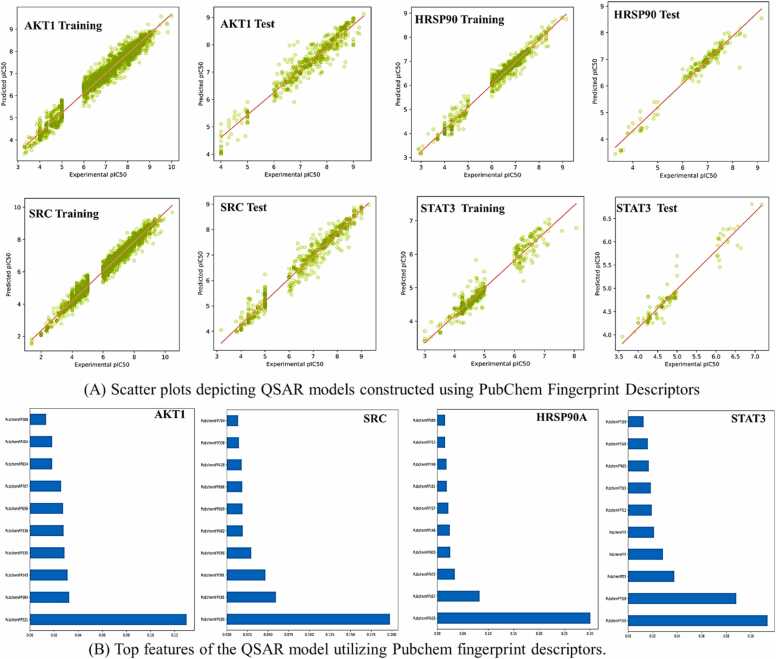
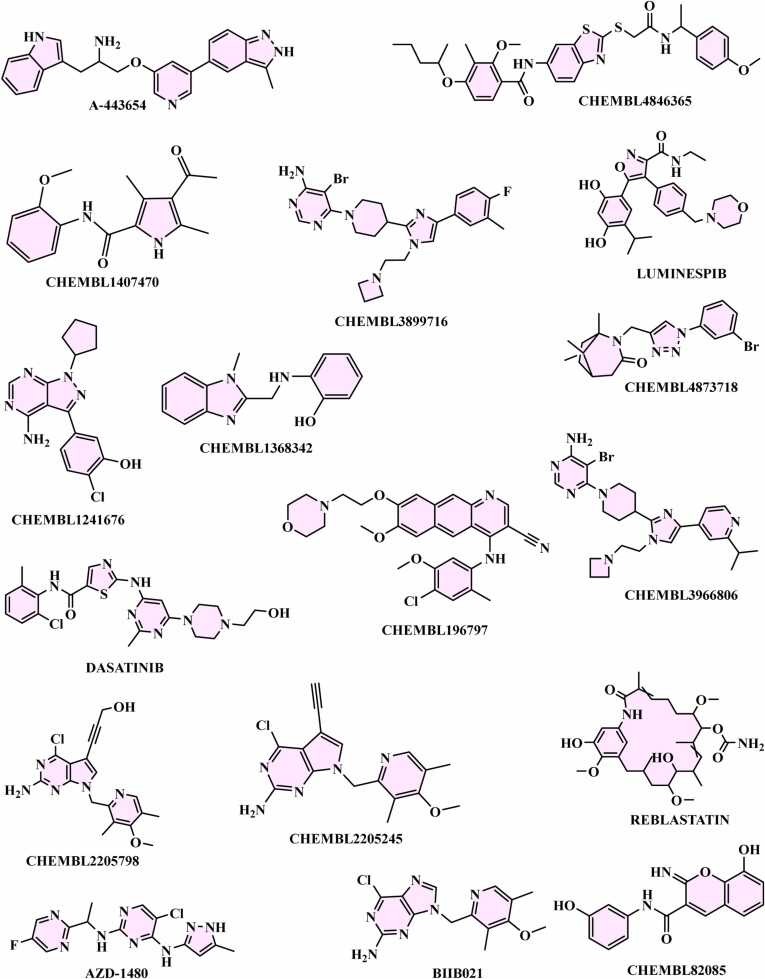
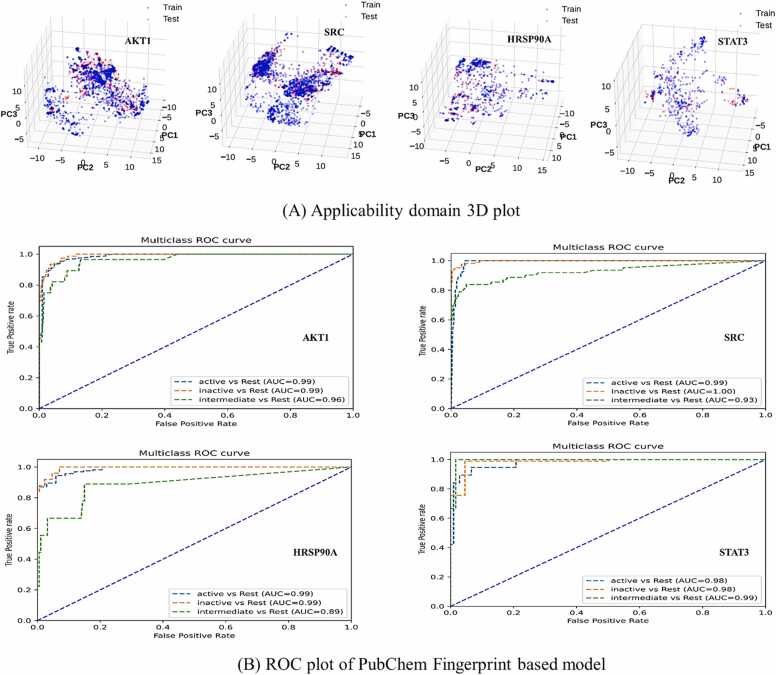
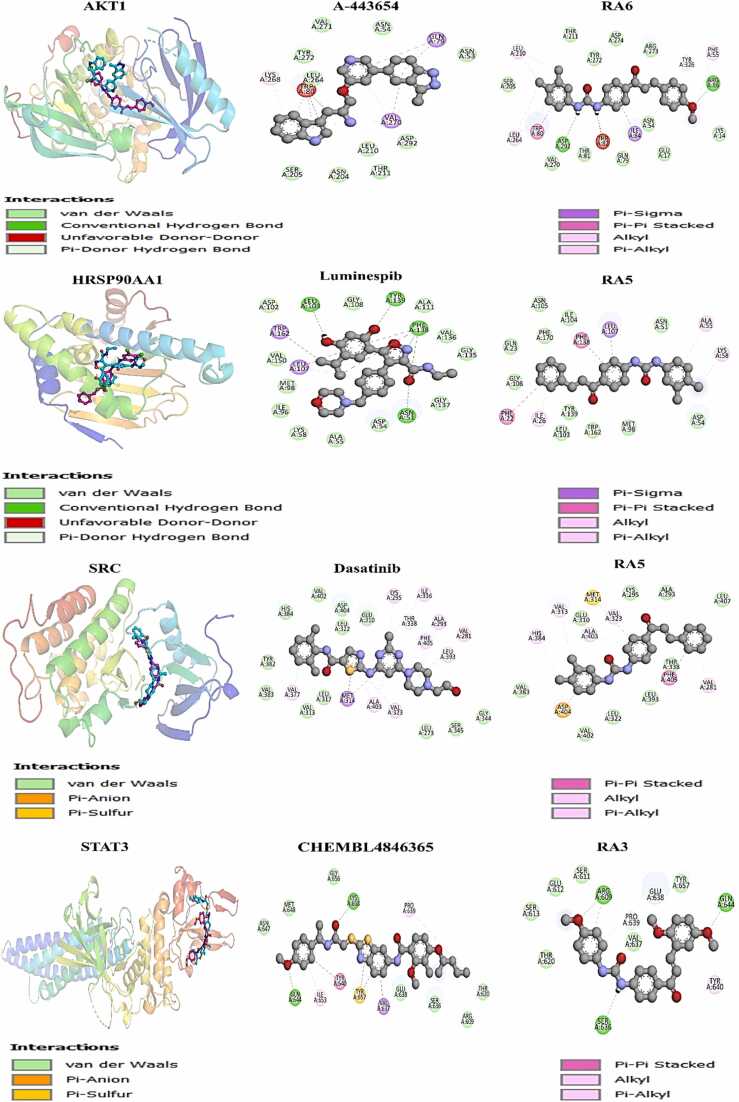
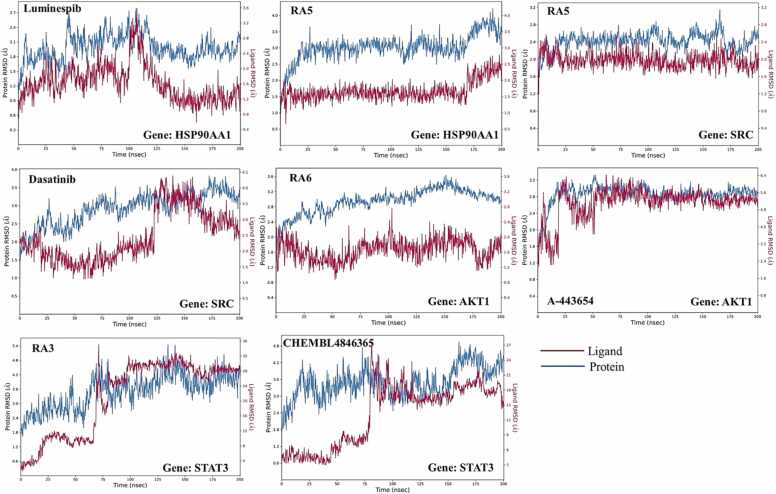
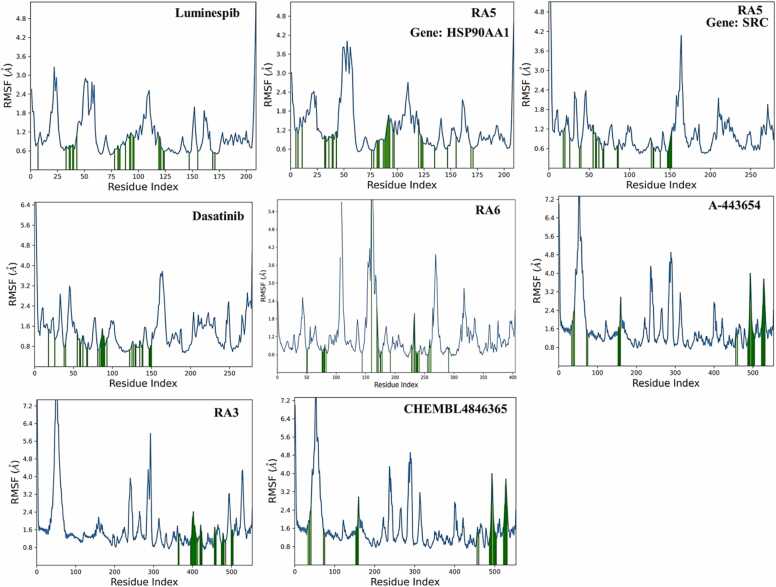
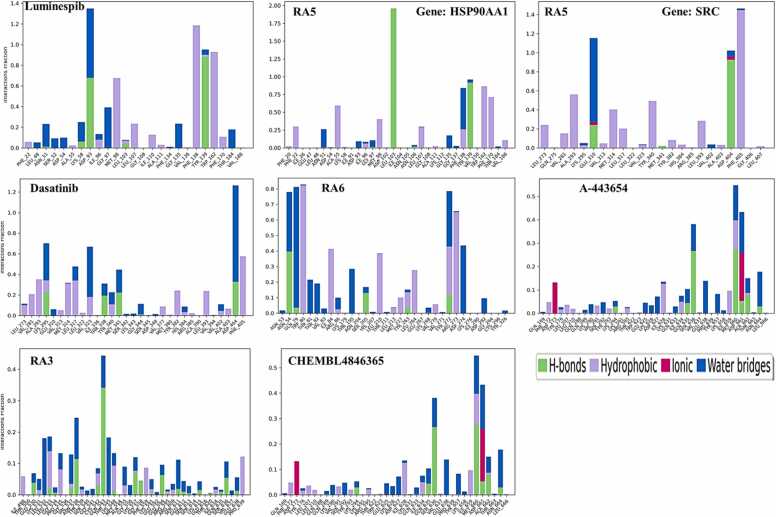
We developed a bio-cheminformatics method, exploring disease inhibition mechanisms using machine learning-enhanced quantitative structure-activity relationship (ML-QSAR) models and knowledge-driven neural networks. ML-QSAR models were developed using molecular fingerprint descriptors and the Random Forest algorithm to explore the chemical spaces of Chalcones inhibitors against diverse disease properties, including antifungal, anti-inflammatory, anticancer, antimicrobial, and antiviral effects. We generated and validated robust machine learning-based bioactivity prediction models (https://github.com/RatulChemoinformatics/QSAR) for the top genes. These models underwent ROC and applicability domain analysis, followed by molecular docking studies to elucidate the molecular mechanisms of the molecules. Through comprehensive neural network analysis, crucial genes such as and were identified. The PubChem fingerprint-based model revealed key descriptors: PubchemFP521 for , PubchemFP180 for , PubchemFP633 for and PubchemFP145 and PubchemFP338 for , consistently contributing to bioactivity across targets. Notably, chalcone derivatives demonstrated significant bioactivity against target genes, with compound RA1 displaying a predictive pIC value of 5.76 against and strong binding affinities across other targets. Compounds RA5 to RA7 also exhibited high binding affinity scores comparable to or exceeding existing drugs. These findings emphasize the importance of knowledge-based neural network-based research for developing effective drugs against diverse disease properties. These interactions warrant further in vitro and in vivo investigations to elucidate their potential in rational drug design. The presented models provide valuable insights for inhibitor design and hold promise for drug development. Future research will prioritize investigating these molecules for , enhancing the comprehension of effectiveness in addressing infectious diseases.
我们开发了一种生物化学信息学方法,利用机器学习增强的定量构效关系(ML-QSAR)模型和知识驱动的神经网络来探索疾病抑制机制。使用分子指纹描述符和随机森林算法开发ML-QSAR模型,以探索查耳酮抑制剂针对多种疾病特性的化学空间,包括抗真菌、抗炎、抗癌、抗菌和抗病毒作用。我们为顶级基因生成并验证了基于机器学习的强大生物活性预测模型(https://github.com/RatulChemoinformatics/QSAR)。这些模型进行了ROC和适用域分析,随后进行分子对接研究以阐明分子机制。通过综合神经网络分析,确定了关键基因如 和 。基于PubChem指纹的模型揭示了关键描述符: 对应的PubchemFP521、 对应的PubchemFP180、 对应的PubchemFP633以及 和 对应的PubchemFP145和PubchemFP338,这些描述符始终对跨靶点的生物活性有贡献。值得注意的是,查耳酮衍生物对靶基因表现出显著的生物活性,化合物RA1对 的预测pIC值为5.76,并且对其他靶点具有强结合亲和力。化合物RA5至RA7也表现出与现有药物相当或超过现有药物的高结合亲和力分数。这些发现强调了基于知识的神经网络研究对于开发针对多种疾病特性的有效药物的重要性。这些相互作用值得进一步进行体外和体内研究,以阐明它们在合理药物设计中的潜力。所提出的模型为抑制剂设计提供了有价值的见解,并为药物开发带来了希望。未来的研究将优先研究这些分子对 的作用,以增强对其在解决传染病方面有效性的理解。