Liyanage Jayamini C, Prendergast Luke, Staudte Robert, De Livera Alysha M
Mathematics and Statistics, School of Computing, Engineering and Mathematical Sciences, La Trobe University, Kingsbury Dr, VIC 3086, Australia.
Bioinformatics. 2024 Jul 25;40(7). doi: 10.1093/bioinformatics/btae470.
Meta-analysis methods widely-used for combining metabolomics data do not account for correlation between metabolites or missing values. Within- and between-study variability are also often overlooked. These can give results with inferior statistical properties, leading to misidentification of biomarkers.
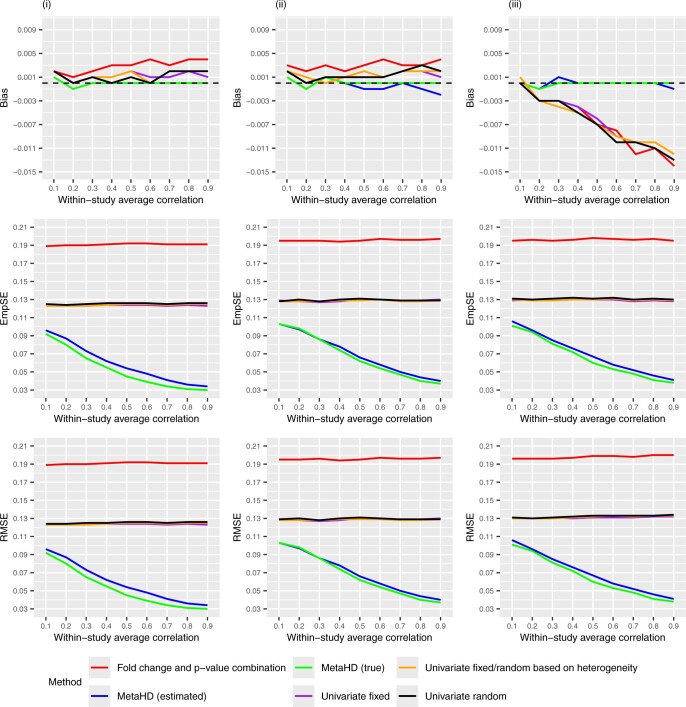
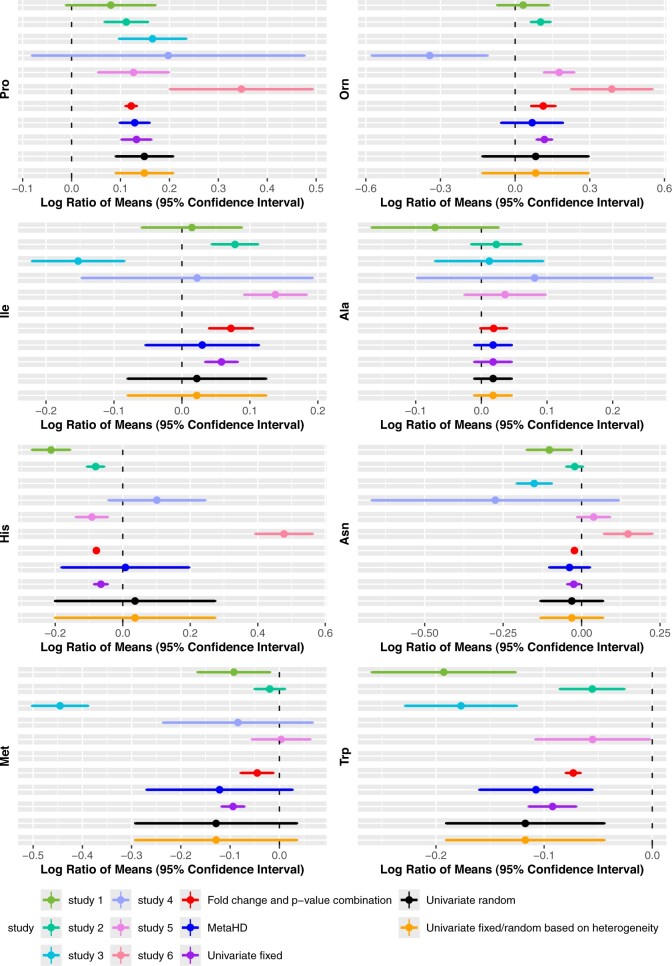
We propose a multivariate meta-analysis model for high-dimensional metabolomics data (MetaHD), which accommodates the correlation between metabolites, within- and between-study variances, and missing values. MetaHD can be used for integrating and collectively analysing individual-level metabolomics data generated from multiple studies as well as for combining summary estimates. We show that MetaHD leads to lower root mean square error compared to the existing approaches. Furthermore, we demonstrate that MetaHD, which exploits the borrowing strength between metabolites, could be particularly useful in the presence of missing data compared to univariate meta-analysis methods, which can return biased estimates in the presence of data missing at random.
The MetaHD R package can be downloaded through Comprehensive R Archive Network (CRAN) repository. A detailed vignette with example datasets and code to prepare data and analyses are available on https://bookdown.org/a2delivera/MetaHD/.
Supplementary data are available at Bioinformatics online.
广泛用于整合代谢组学数据的荟萃分析方法未考虑代谢物之间的相关性或缺失值。研究内和研究间的变异性也常常被忽视。这些可能会导致统计特性较差的结果,从而导致生物标志物的误识别。
我们提出了一种用于高维代谢组学数据的多变量荟萃分析模型(MetaHD),该模型考虑了代谢物之间的相关性、研究内和研究间的方差以及缺失值。MetaHD可用于整合和集体分析来自多项研究的个体水平代谢组学数据,以及用于合并汇总估计值。我们表明,与现有方法相比,MetaHD导致更低的均方根误差。此外,我们证明,与单变量荟萃分析方法相比,利用代谢物之间的借用强度的MetaHD在存在缺失数据的情况下可能特别有用,单变量荟萃分析方法在存在随机缺失数据的情况下可能会返回有偏差的估计值。
MetaHD R包可以通过综合R存档网络(CRAN)存储库下载。在https://bookdown.org/a2delivera/MetaHD/上提供了带有示例数据集以及准备数据和分析的代码的详细 vignette。
补充数据可在《生物信息学》在线获取。