Department of Creative Informatics, Graduate School of Information Science and Technology, The University of Tokyo, Bunkyo, Tokyo, Japan.
Bioinformation and DDBJ Center, National Institute of Genetics, Mishima, Shizuoka, Japan.
F1000Res. 2024 Jun 24;11:889. doi: 10.12688/f1000research.122924.2. eCollection 2022.
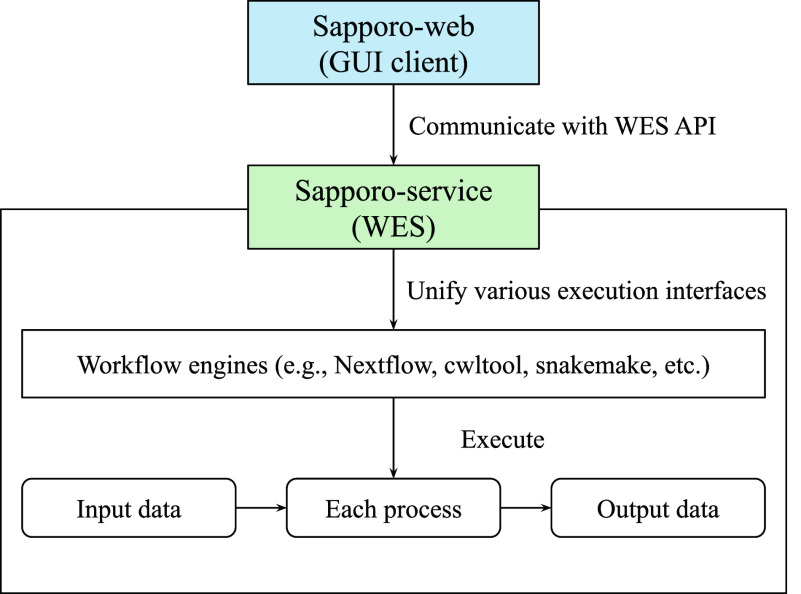
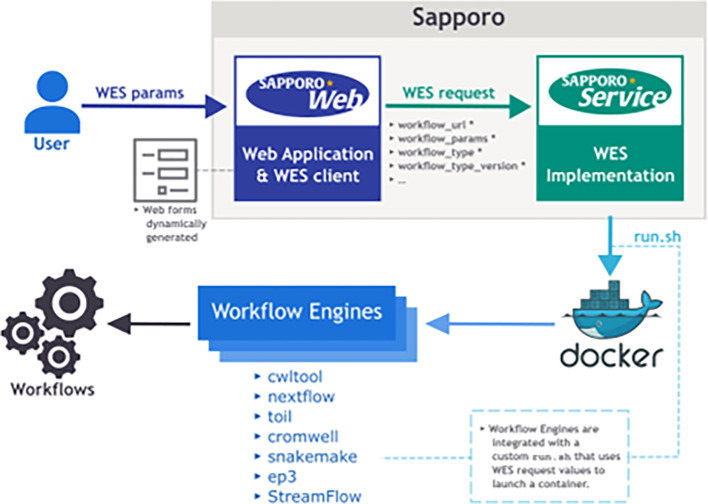
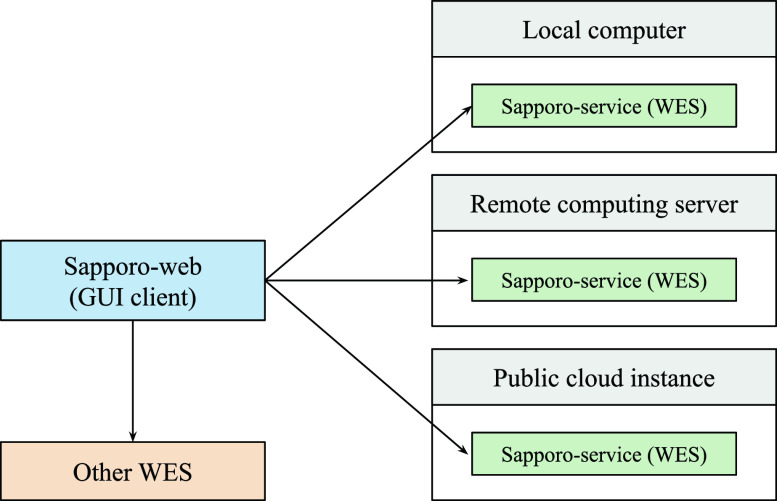
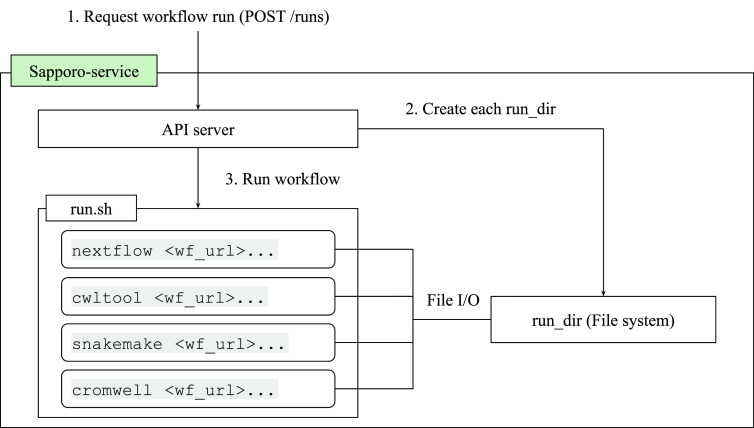
The increased demand for efficient computation in data analysis encourages researchers in biomedical science to use workflow systems. Workflow systems, or so-called workflow languages, are used for the description and execution of a set of data analysis steps. Workflow systems increase the productivity of researchers, specifically in fields that use high-throughput DNA sequencing applications, where scalable computation is required. As systems have improved the portability of data analysis workflows, research communities are able to share workflows to reduce the cost of building ordinary analysis procedures. However, having multiple workflow systems in a research field has resulted in the distribution of efforts across different workflow system communities. As each workflow system has its unique characteristics, it is not feasible to learn every single system in order to use publicly shared workflows. Thus, we developed Sapporo, an application to provide a unified layer of workflow execution upon the differences of various workflow systems. Sapporo has two components: an application programming interface (API) that receives the request of a workflow run and a browser-based client for the API. The API follows the Workflow Execution Service API standard proposed by the Global Alliance for Genomics and Health. The current implementation supports the execution of workflows in four languages: Common Workflow Language, Workflow Description Language, Snakemake, and Nextflow. With its extensible and scalable design, Sapporo can support the research community in utilizing valuable resources for data analysis.
数据分析中对高效计算的需求增加,促使生物医学科学研究人员使用工作流程系统。工作流程系统,或所谓的工作流程语言,用于描述和执行一组数据分析步骤。工作流程系统提高了研究人员的工作效率,特别是在需要可扩展计算的高通量 DNA 测序应用领域。随着系统提高了数据分析工作流程的可移植性,研究社区能够共享工作流程,以降低构建普通分析程序的成本。然而,在一个研究领域中存在多个工作流程系统,导致在不同的工作流程系统社区中分散了精力。由于每个工作流程系统都有其独特的特点,不可能为了使用公共共享的工作流程而去学习每个单独的系统。因此,我们开发了 Sapporo,这是一种在各种工作流程系统的差异之上提供统一的工作流程执行层的应用程序。Sapporo 有两个组件:一个接收工作流程运行请求的应用程序编程接口 (API),以及一个基于浏览器的 API 客户端。API 遵循由全球基因组和健康联盟提出的工作流程执行服务 API 标准。当前的实现支持四种语言的工作流程执行:通用工作流程语言、工作流程描述语言、Snakemake 和 Nextflow。凭借其可扩展和可扩展的设计,Sapporo 可以支持研究社区利用数据分析的宝贵资源。