Davison Jack R, Hadjithomas Michalis, Romeril Stuart P, Choi Yoon Jong, Bentley Keith W, Biggins John B, Chacko Nadia, Castaldi M Paola, Chan Lawrence K, Cumming Jared N, Downes Thomas D, Eisenhauer Eric L, Fei Fan, Fontaine Benjamin M, Endalur Gopinarayanan Venkatesh, Gurnani Srishti, Hecht Audrey, Hosford Christopher J, Ibrahim Ashraf, Jagels Annika, Joubran Camil, Kim Ji-Nu, Lisher John P, Liu Daniel D, Lyles James T, Mannara Matteo N, Murray Gordon J, Musial Emilia, Niu Mengyao, Olivares-Amaya Roberto, Percuoco Marielle, Saalau Susanne, Sharpe Kristen, Sheahan Anjali V, Thevakumaran Neroshan, Thompson James E, Thompson Dawn A, Wiest Aric, Wyka Stephen A, Yano Jason, Verdine Gregory L
LifeMine Therapeutics, 30 Acorn Park Drive, Cambridge, Massachusetts 02140, United States.
Departments of Chemistry and Chemical Biology, and Stem Cell and Regenerative Biology, Harvard University and Harvard Medical School, 12 Oxford Street, Cambridge, Massachusetts 02138, United States.
J Med Chem. 2024 Aug 8;67(15):13147-13173. doi: 10.1021/acs.jmedchem.4c01095. Epub 2024 Jul 30.
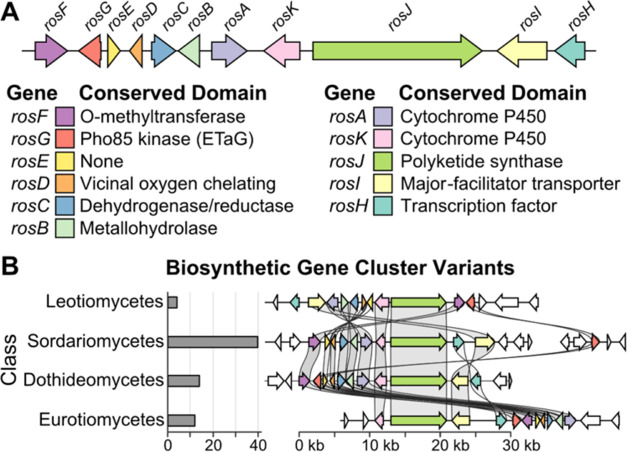
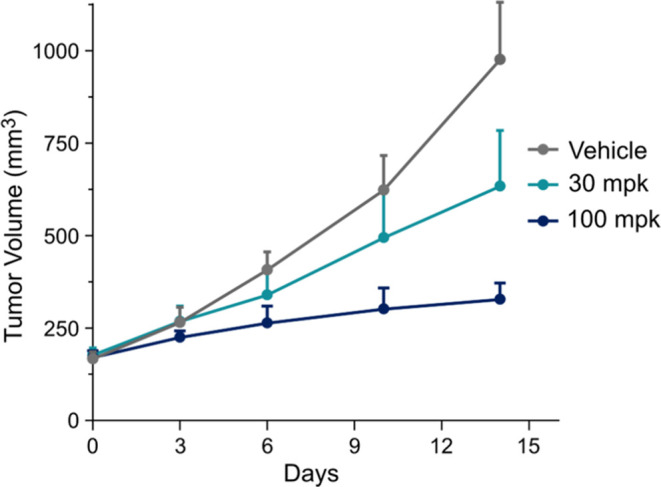
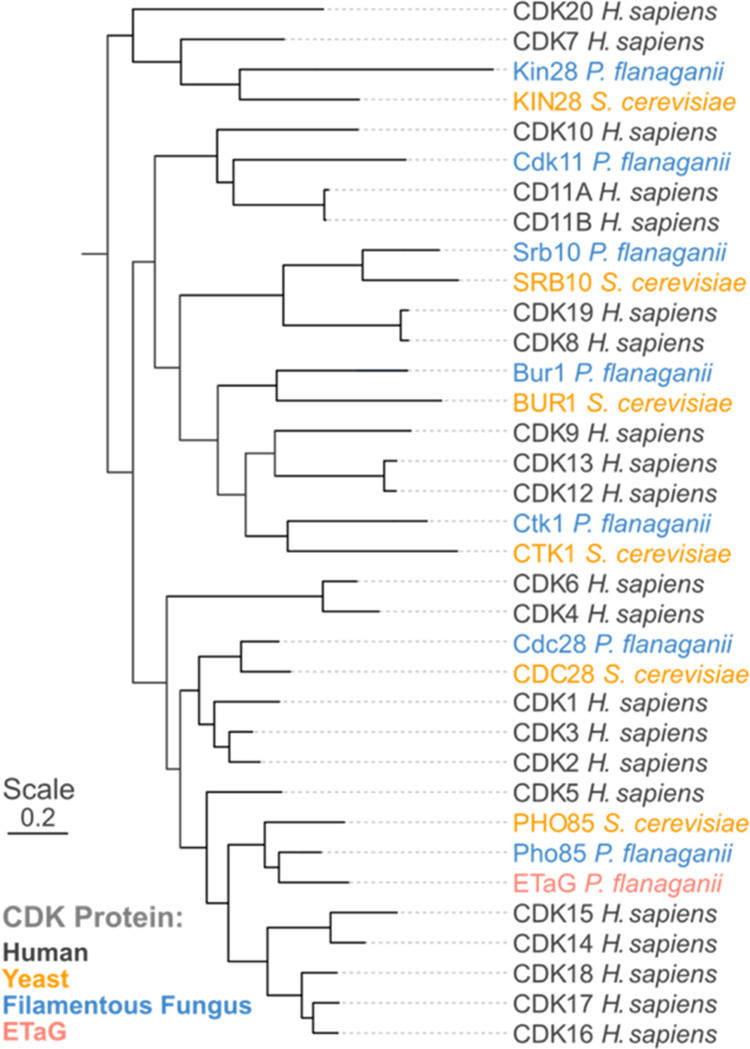
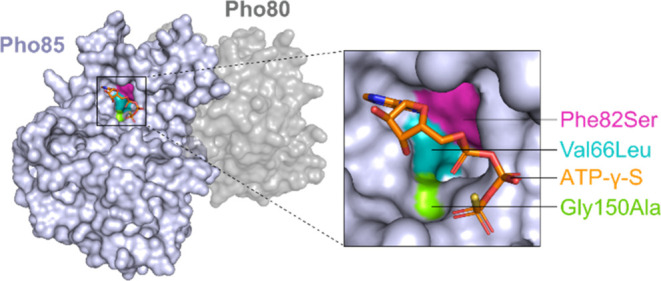
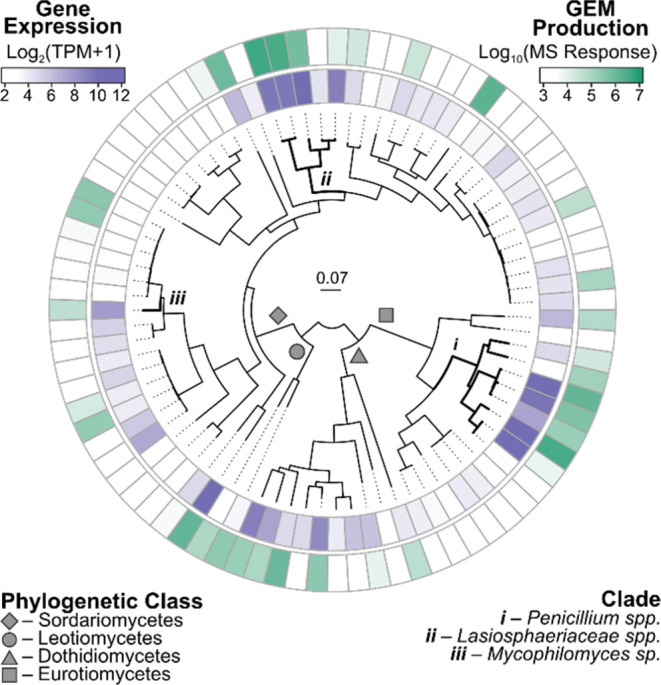
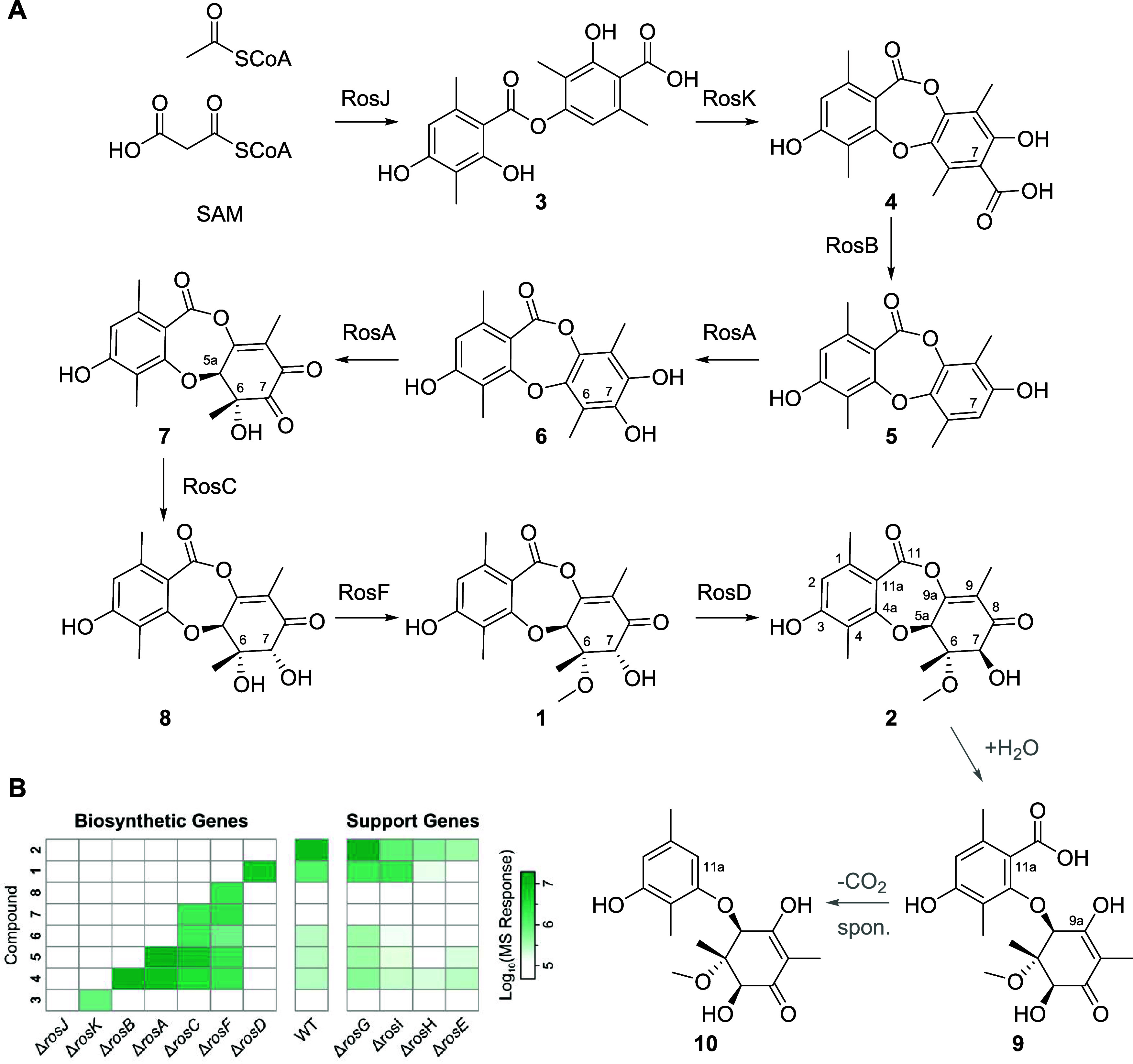
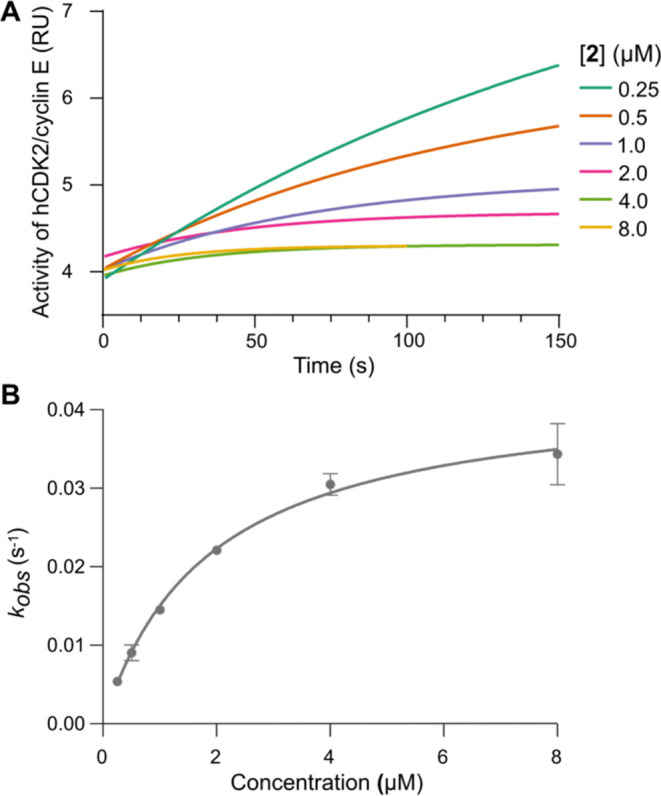
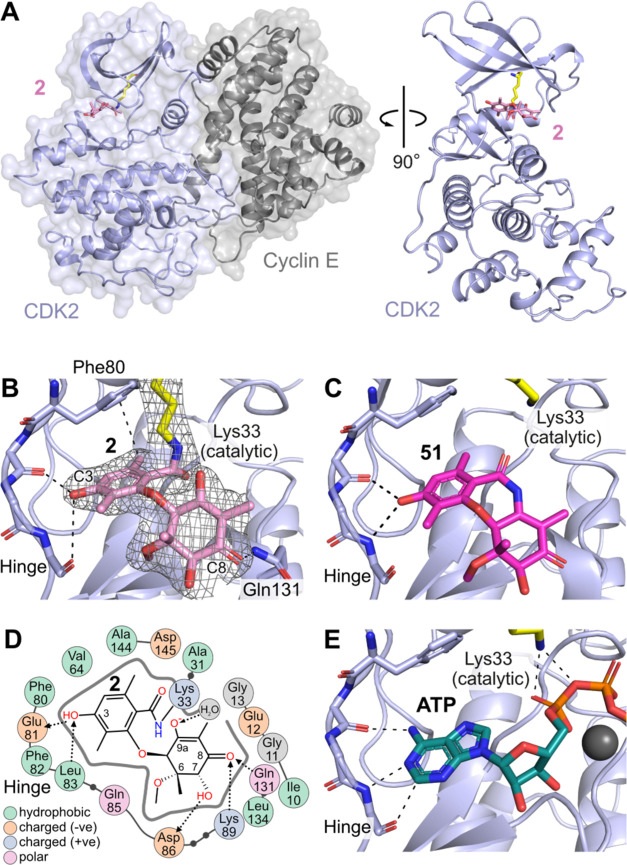
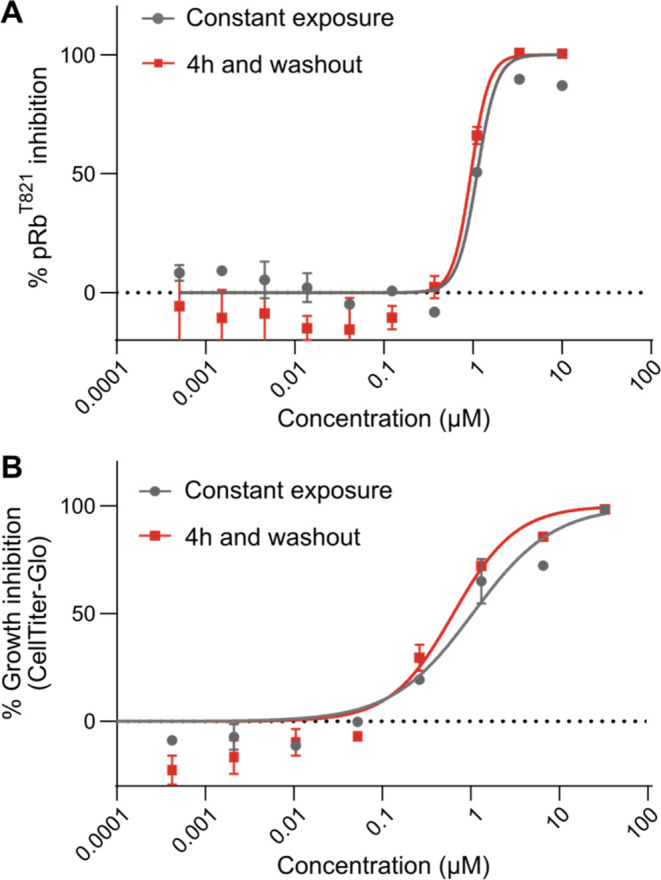
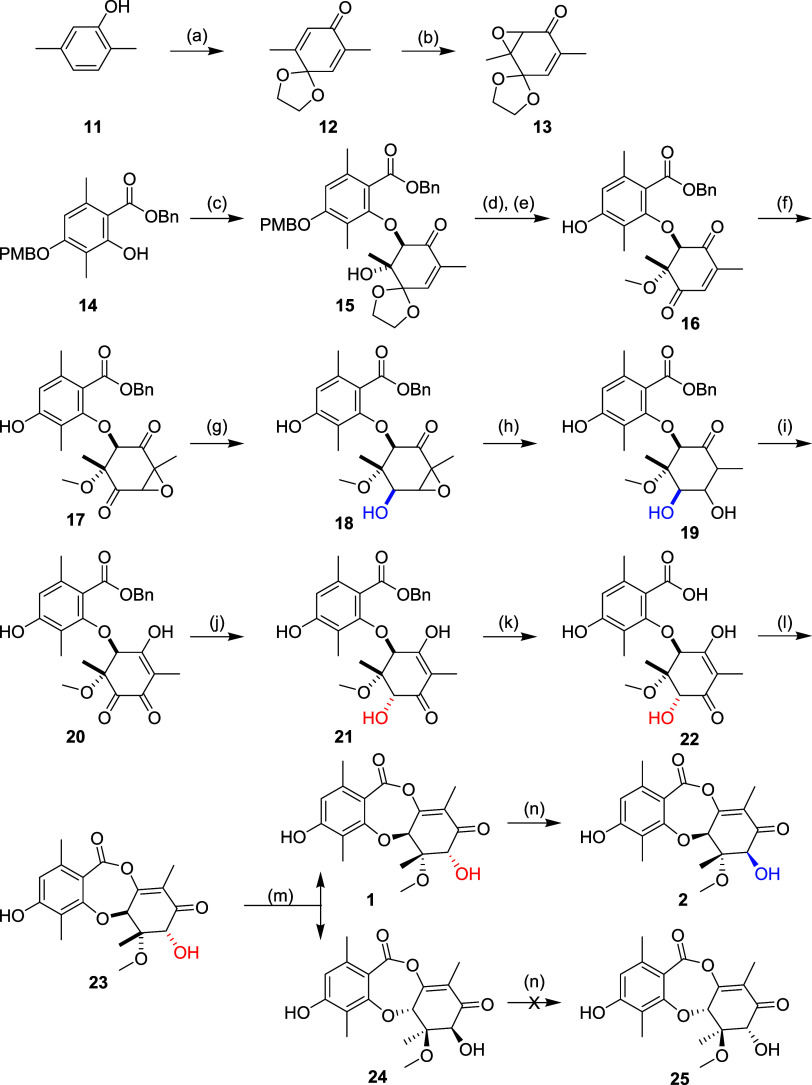
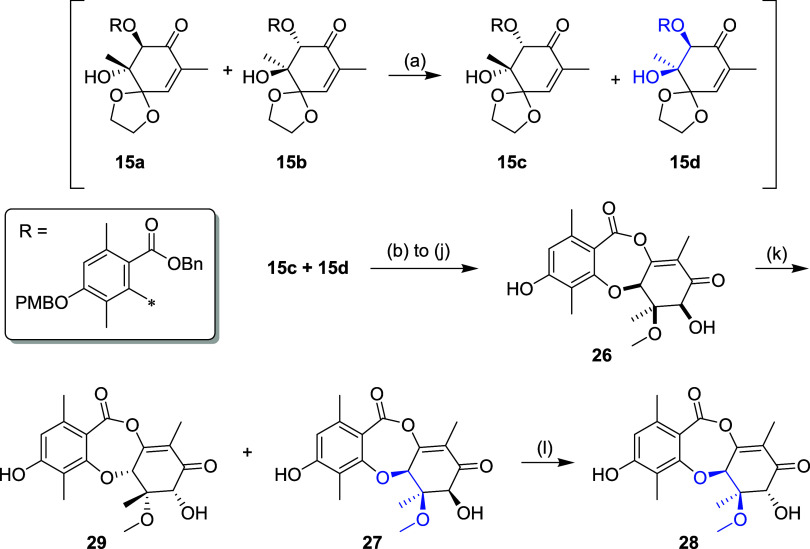
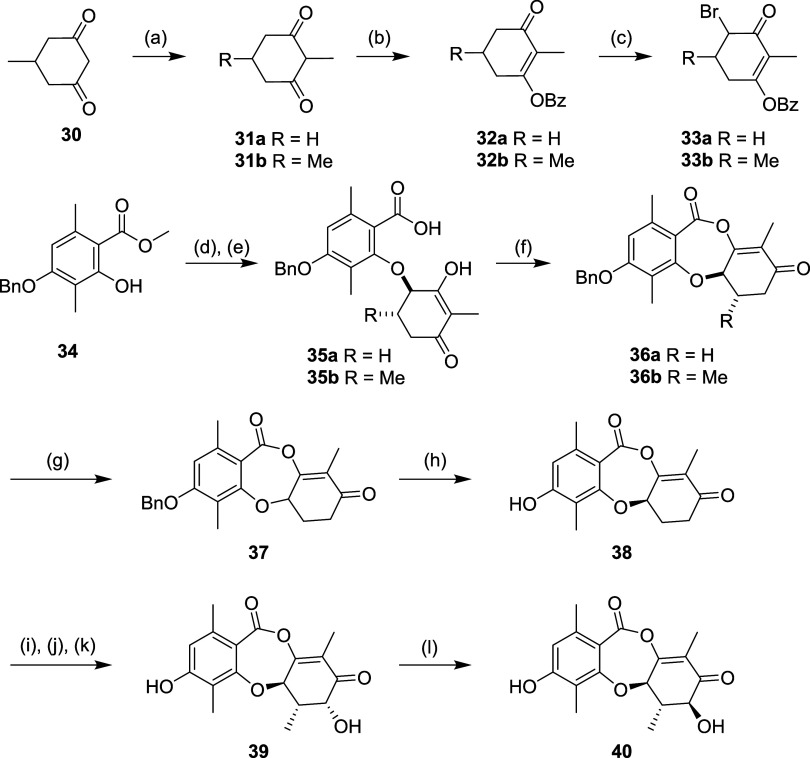
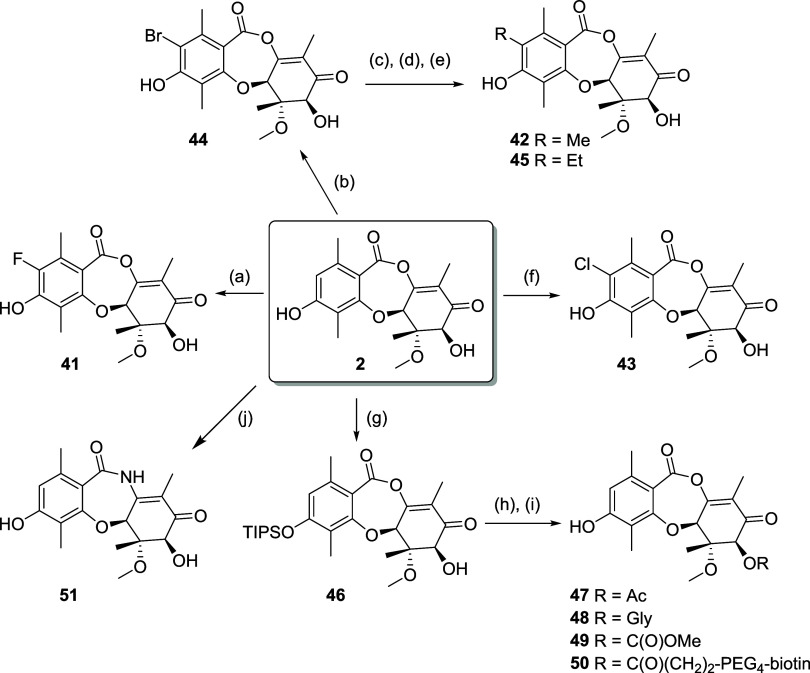
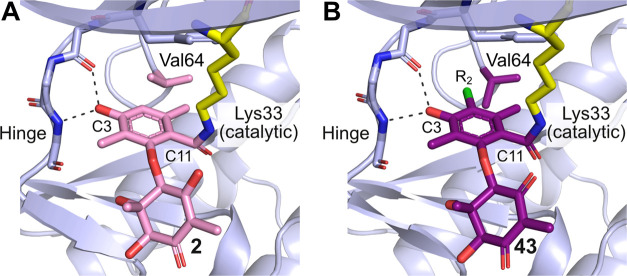
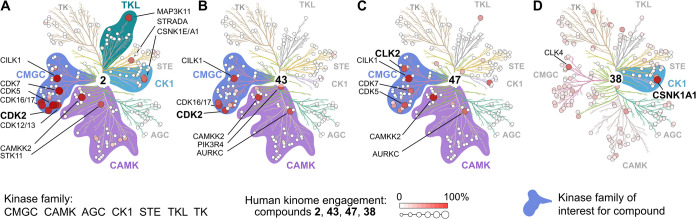
Fungi have historically been the source of numerous important medicinal compounds, but full exploitation of their genetic potential for drug development has been hampered in traditional discovery paradigms. Here we describe a radically different approach, top-down drug discovery (TD), starting with a massive digital search through a database of over 100,000 fully genomicized fungi to identify loci encoding molecules with a predetermined human target. We exemplify TD by the selection of cyclin-dependent kinases (CDKs) as targets and the discovery of two molecules, and , which inhibit therapeutically important human CDKs. and exhibit a remarkable mechanism, forming a site-selective covalent bond to the CDK active site Lys. We explored the structure-activity relationship via semi- and total synthesis, generating an analog, , with improved kinase selectivity, bioavailability, and efficacy. This work highlights the power of TD to identify mechanistically and structurally novel molecules for the development of new medicines.
从历史上看,真菌一直是众多重要药用化合物的来源,但在传统的发现模式中,其药物开发的遗传潜力尚未得到充分挖掘。在此,我们描述了一种截然不同的方法——自上而下的药物发现(TD),即首先通过对一个包含超过100,000种全基因组真菌的数据库进行大规模数字搜索,以识别编码具有预定人类靶点分子的基因座。我们以细胞周期蛋白依赖性激酶(CDK)作为靶点,通过TD发现了两种分子,即[具体分子名称1]和[具体分子名称2],它们可抑制具有重要治疗意义的人类CDK。[具体分子名称1]和[具体分子名称2]展现出一种独特的机制,与CDK活性位点的赖氨酸形成位点选择性共价键。我们通过半合成和全合成探索了构效关系,生成了一种具有改善的激酶选择性、生物利用度和功效的类似物[具体分子名称3]。这项工作凸显了TD在识别用于新药开发的具有独特作用机制和结构的分子方面的强大能力。