Institute of Structural and Molecular Biology, University College London, Gower Street, London, WC1E 6BT, UK.
Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Pawinskiego 5a, 02-106, Warsaw, Poland.
Sci Rep. 2024 Aug 5;14(1):18149. doi: 10.1038/s41598-024-68468-7.
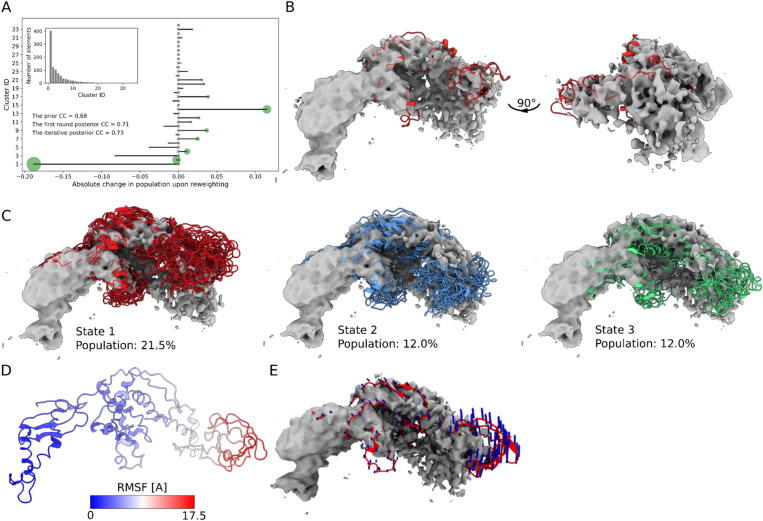
Cryogenic electron microscopy (cryo-EM) has emerged as a powerful method for the determination of structures of complex biological molecules. The accurate characterisation of the dynamics of such systems, however, remains a challenge. To address this problem, we introduce cryoENsemble, a method that applies Bayesian reweighting to conformational ensembles derived from molecular dynamics simulations to improve their agreement with cryo-EM data, thus enabling the extraction of dynamics information. We illustrate the use of cryoENsemble to determine the dynamics of the ribosome-bound state of the co-translational chaperone trigger factor (TF). We also show that cryoENsemble can assist with the interpretation of low-resolution, noisy or unaccounted regions of cryo-EM maps. Notably, we are able to link an unaccounted part of the cryo-EM map to the presence of another protein (methionine aminopeptidase, or MetAP), rather than to the dynamics of TF, and model its TF-bound state. Based on these results, we anticipate that cryoENsemble will find use for challenging heterogeneous cryo-EM maps for biomolecular systems encompassing dynamic components.
低温电子显微镜(cryo-EM)已成为确定复杂生物分子结构的强大方法。然而,此类系统动态的准确描述仍然是一个挑战。为了解决这个问题,我们引入了 cryoENsemble 方法,该方法将贝叶斯重新加权应用于从分子动力学模拟中得出的构象集合,以提高它们与低温电子显微镜数据的一致性,从而能够提取动态信息。我们说明了 cryoENsemble 在确定共翻译伴侣触发因子(TF)结合核糖体的状态的动力学方面的用途。我们还表明,cryoENsemble 可以帮助解释低温电子显微镜图谱中低分辨率、嘈杂或未解释的区域。值得注意的是,我们能够将低温电子显微镜图谱中未解释的部分与另一种蛋白质(甲硫氨酸氨肽酶或 MetAP)的存在联系起来,而不是与 TF 的动态联系起来,并对其 TF 结合状态进行建模。基于这些结果,我们预计 cryoENsemble 将适用于包含动态成分的生物分子系统具有挑战性的异质低温电子显微镜图谱。