InstaDeep, Cambridge, MA 02142, United States.
InstaDeep, Paris 75010, France.
Bioinformatics. 2024 Sep 2;40(9). doi: 10.1093/bioinformatics/btae529.
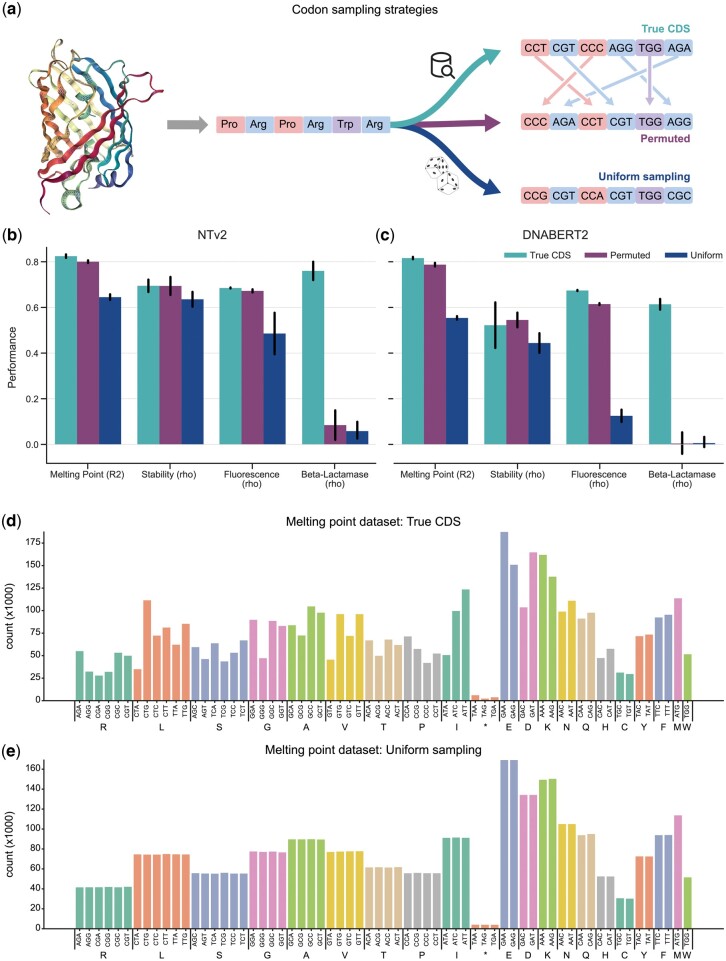
Large language models, trained on enormous corpora of biological sequences, are state-of-the-art for downstream genomic and proteomic tasks. Since the genome contains the information to encode all proteins, genomic language models (gLMs) hold the potential to make downstream predictions not only about DNA sequences, but also about proteins. However, the performance of gLMs on protein tasks remains unknown, due to few tasks pairing proteins with the coding DNA sequences (CDS) that can be processed by gLMs.
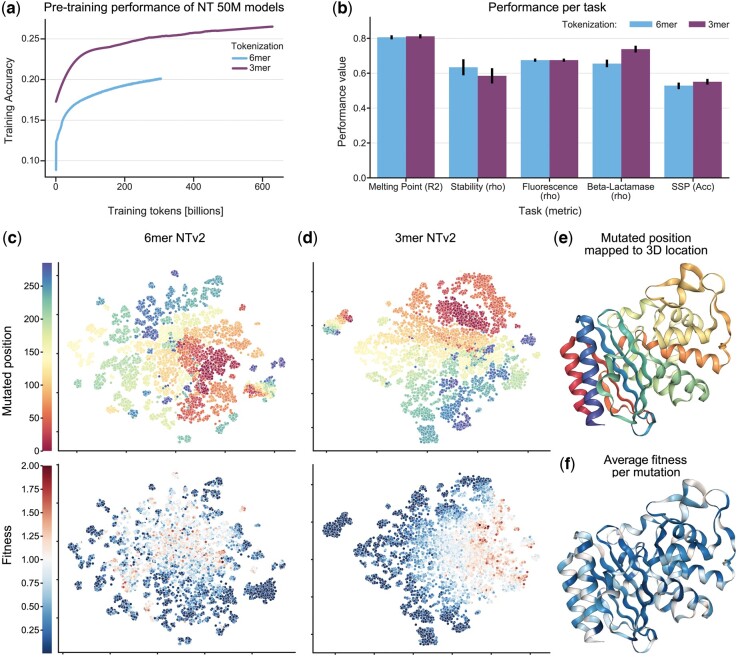
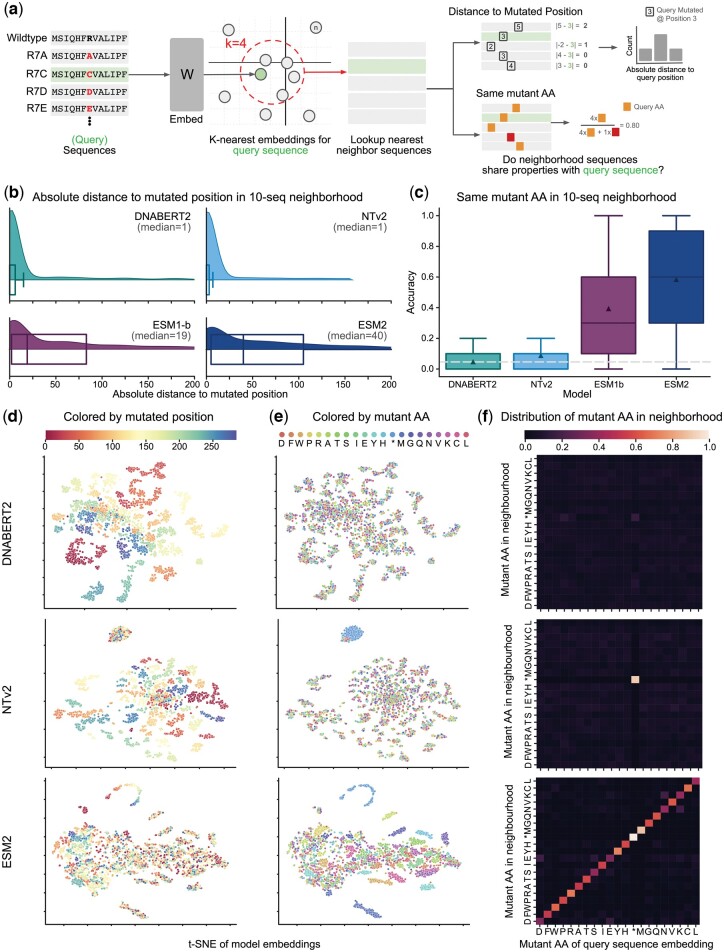
In this work, we curated five such datasets and used them to evaluate the performance of gLMs and proteomic language models (pLMs). We show that gLMs are competitive and even outperform their pLMs counterparts on some tasks. The best performance was achieved using the retrieved CDS compared to sampling strategies. We found that training a joint genomic-proteomic model outperforms each individual approach, showing that they capture different but complementary sequence representations, as we demonstrate through model interpretation of their embeddings. Lastly, we explored different genomic tokenization schemes to improve downstream protein performance. We trained a new Nucleotide Transformer (50M) foundation model with 3mer tokenization that outperforms its 6mer counterpart on protein tasks while maintaining performance on genomics tasks. The application of gLMs to proteomics offers the potential to leverage rich CDS data, and in the spirit of the central dogma, the possibility of a unified and synergistic approach to genomics and proteomics.
We make our inference code, 3mer pre-trained model weights and datasets available.
在大量生物序列语料库上进行训练的大型语言模型,是用于下游基因组和蛋白质组任务的最新技术。由于基因组包含了编码所有蛋白质的信息,因此基因组语言模型(gLMs)有可能不仅对 DNA 序列,而且对蛋白质进行下游预测。然而,由于很少有任务将蛋白质与可由 gLMs 处理的编码 DNA 序列(CDS)配对,因此 gLMs 在蛋白质任务上的性能仍然未知。
在这项工作中,我们整理了五个这样的数据集,并使用它们来评估 gLMs 和蛋白质语言模型(pLMs)的性能。我们表明 gLMs 在某些任务上具有竞争力,甚至优于它们的 pLMs 对应物。与采样策略相比,使用检索到的 CDS 获得了最佳性能。我们发现,训练一个联合基因组-蛋白质模型优于每个单独的方法,这表明它们捕获了不同但互补的序列表示,正如我们通过对其嵌入的模型解释来证明的那样。最后,我们探索了不同的基因组标记方案,以提高下游蛋白质性能。我们使用 3mer 标记化训练了一个新的核苷酸转换器(50M)基础模型,该模型在蛋白质任务上的表现优于其 6mer 对应物,同时保持了基因组任务的性能。gLMs 在蛋白质组学中的应用有可能利用丰富的 CDS 数据,并且本着中心法则的精神,有可能采用一种统一的、协同的基因组学和蛋白质组学方法。
我们提供推理代码、3mer 预训练模型权重和数据集。