Tata Institute of Fundamental Research Hyderabad, Telangana 500046, India.
Nucleic Acids Res. 2024 Oct 14;52(18):10836-10849. doi: 10.1093/nar/gkae749.
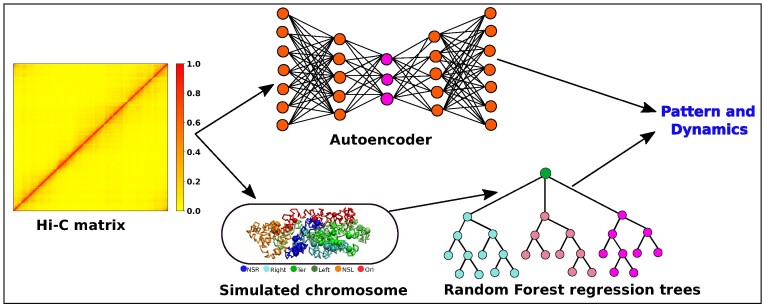
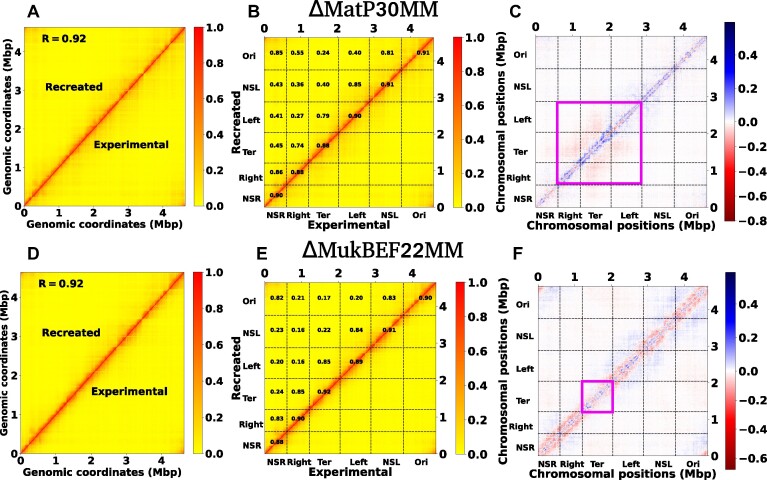
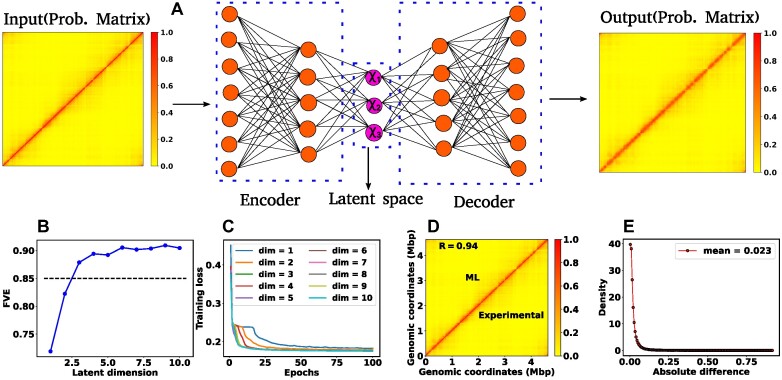
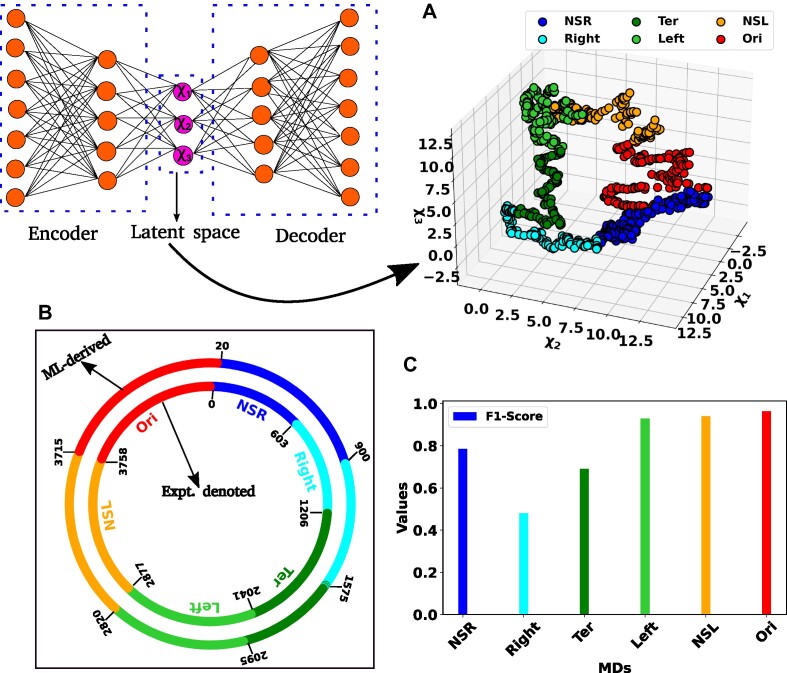
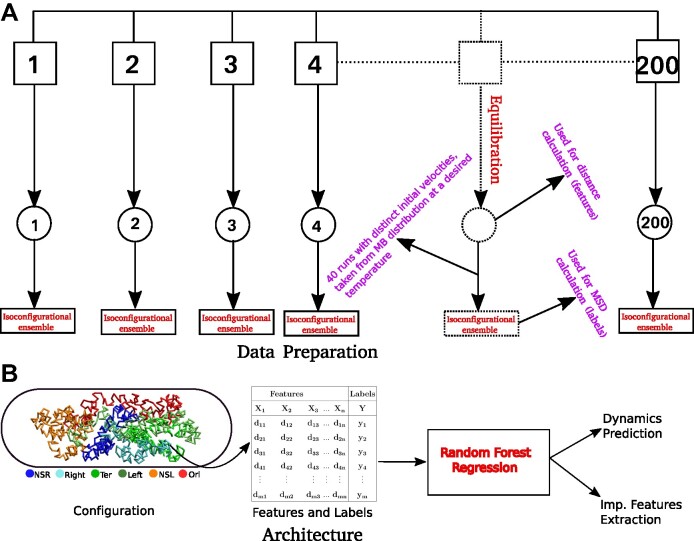
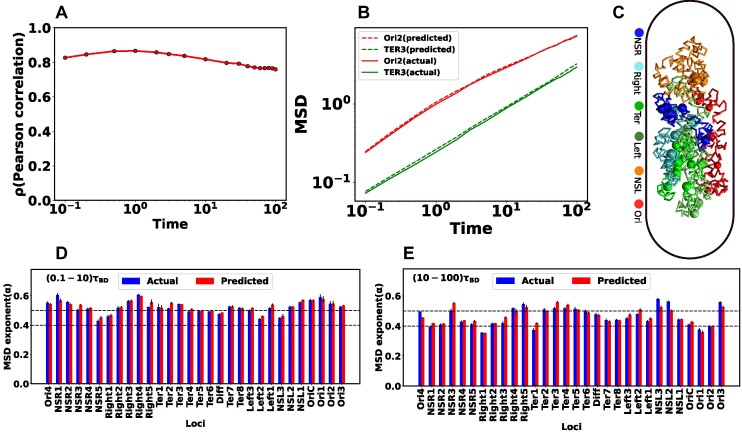
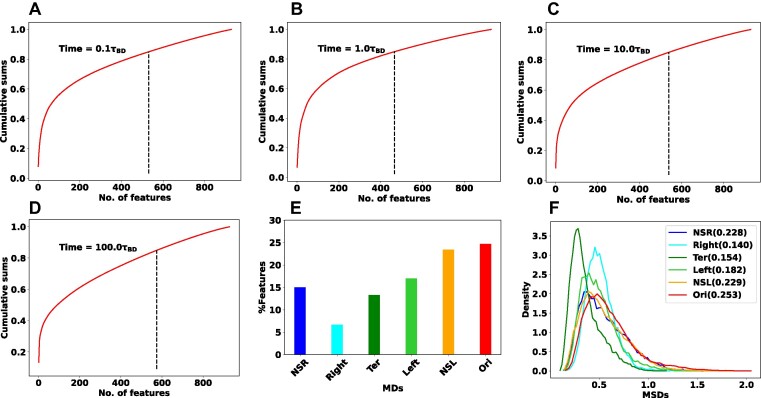
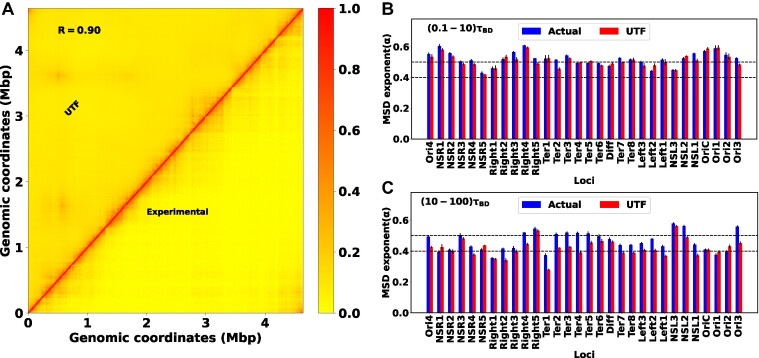
High dimensional nature of the chromosomal conformation contact map ('Hi-C Map'), even for microscopically small bacterial cell, poses challenges for extracting meaningful information related to its complex organization. Here we first demonstrate that an artificial deep neural network-based machine-learnt (ML) low-dimensional representation of a recently reported Hi-C interaction map of archetypal bacteria Escherichia coli can decode crucial underlying structural pattern. The ML-derived representation of Hi-C map can automatically detect a set of spatially distinct domains across E. coli genome, sharing reminiscences of six putative macro-domains previously posited via recombination assay. Subsequently, a ML-generated model assimilates the intricate relationship between large array of Hi-C-derived chromosomal contact probabilities and respective diffusive dynamics of each individual chromosomal gene and identifies an optimal number of functionally important chromosomal contact-pairs that are majorly responsible for heterogenous, coordinate-dependent sub-diffusive motions of chromosomal loci. Finally, the ML models, trained on wild-type E. coli show-cased its predictive capabilities on mutant bacterial strains, shedding light on the structural and dynamic nuances of ΔMatP30MM and ΔMukBEF22MM chromosomes. Overall our results illuminate the power of ML techniques in unraveling the complex relationship between structure and dynamics of bacterial chromosomal loci, promising meaningful connections between ML-derived insights and biological phenomena.
高维的染色体构象接触图谱(“Hi-C 图谱”)性质,即使对于微观的细菌细胞,也给提取与其复杂组织相关的有意义信息带来了挑战。在这里,我们首先证明,基于人工深度神经网络的机器学习(ML)可以对最近报道的典型细菌大肠杆菌的 Hi-C 相互作用图谱进行低维表示,从而解码关键的潜在结构模式。ML 衍生的 Hi-C 图谱表示可以自动检测大肠杆菌基因组中一组空间上不同的区域,这些区域共享先前通过重组试验提出的六个假定的大型结构域的特征。随后,一个 ML 生成的模型综合了大量的 Hi-C 衍生的染色体接触概率与每个染色体基因的扩散动力学之间的复杂关系,并确定了一组功能上重要的染色体接触对的最佳数量,这些接触对主要负责染色体位置的异质、协调相关的亚扩散运动。最后,在野生型大肠杆菌上训练的 ML 模型展示了其在突变细菌株上的预测能力,揭示了ΔMatP30MM 和ΔMukBEF22MM 染色体的结构和动态细微差别。总的来说,我们的结果阐明了 ML 技术在揭示细菌染色体位置的结构和动力学之间复杂关系方面的强大功能,为 ML 衍生的见解与生物学现象之间的有意义联系铺平了道路。