Department of Neurology, University of California, San Francisco, CA 94143.
UCSF Weill Institute for Neurosciences, San Francisco, CA 94143.
Proc Natl Acad Sci U S A. 2024 Sep 10;121(37):e2319804121. doi: 10.1073/pnas.2319804121. Epub 2024 Sep 3.
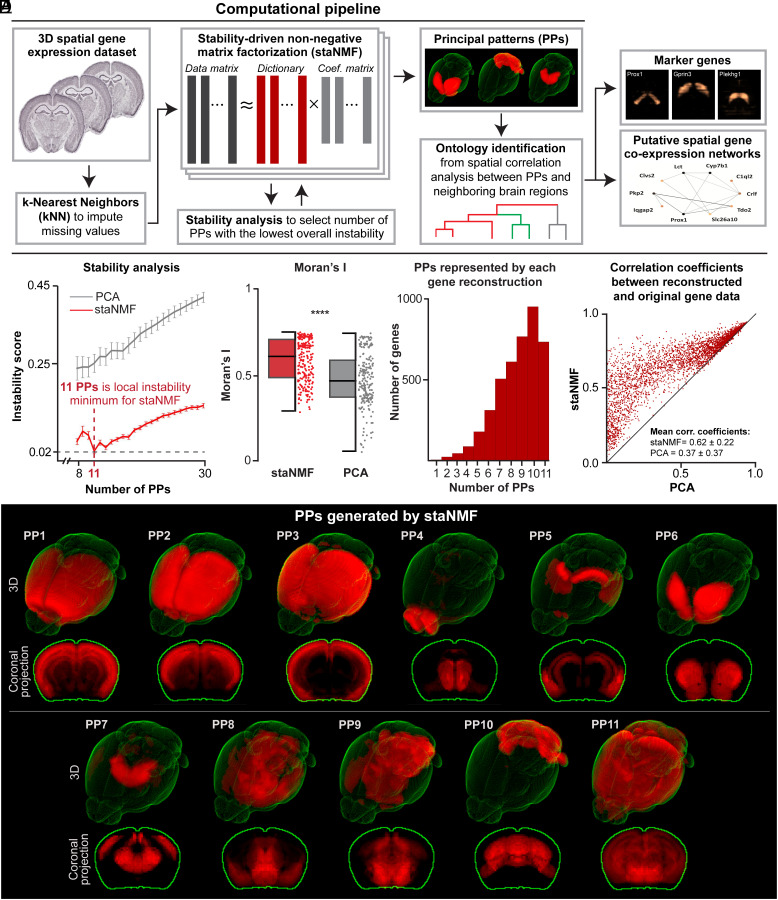
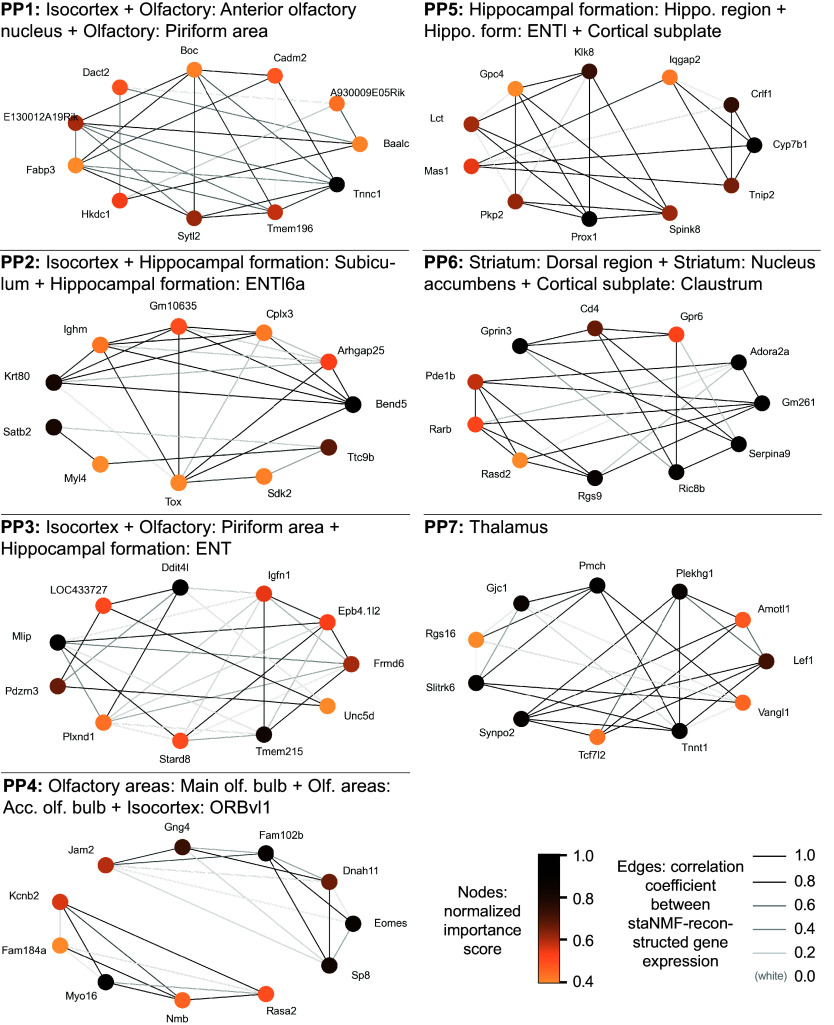
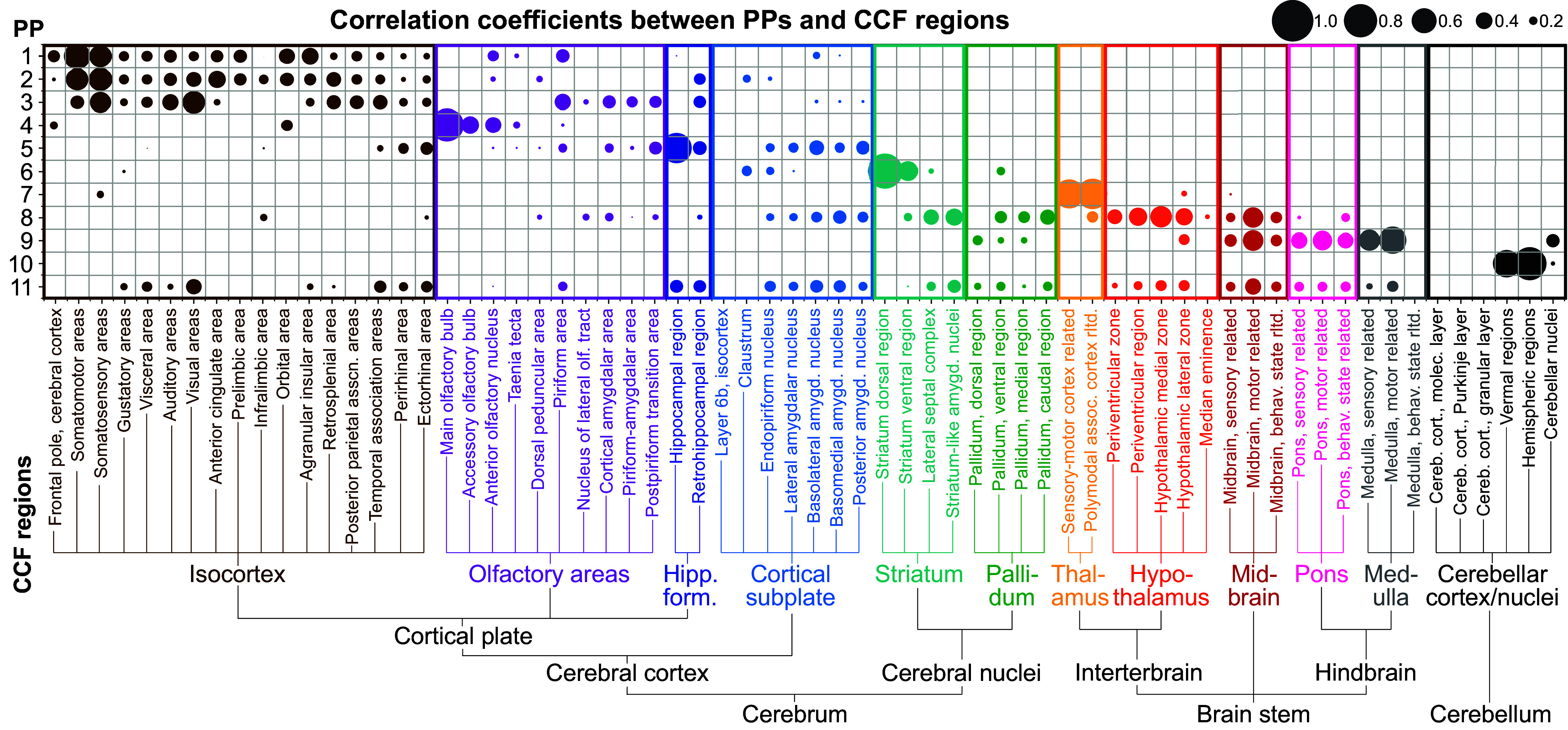
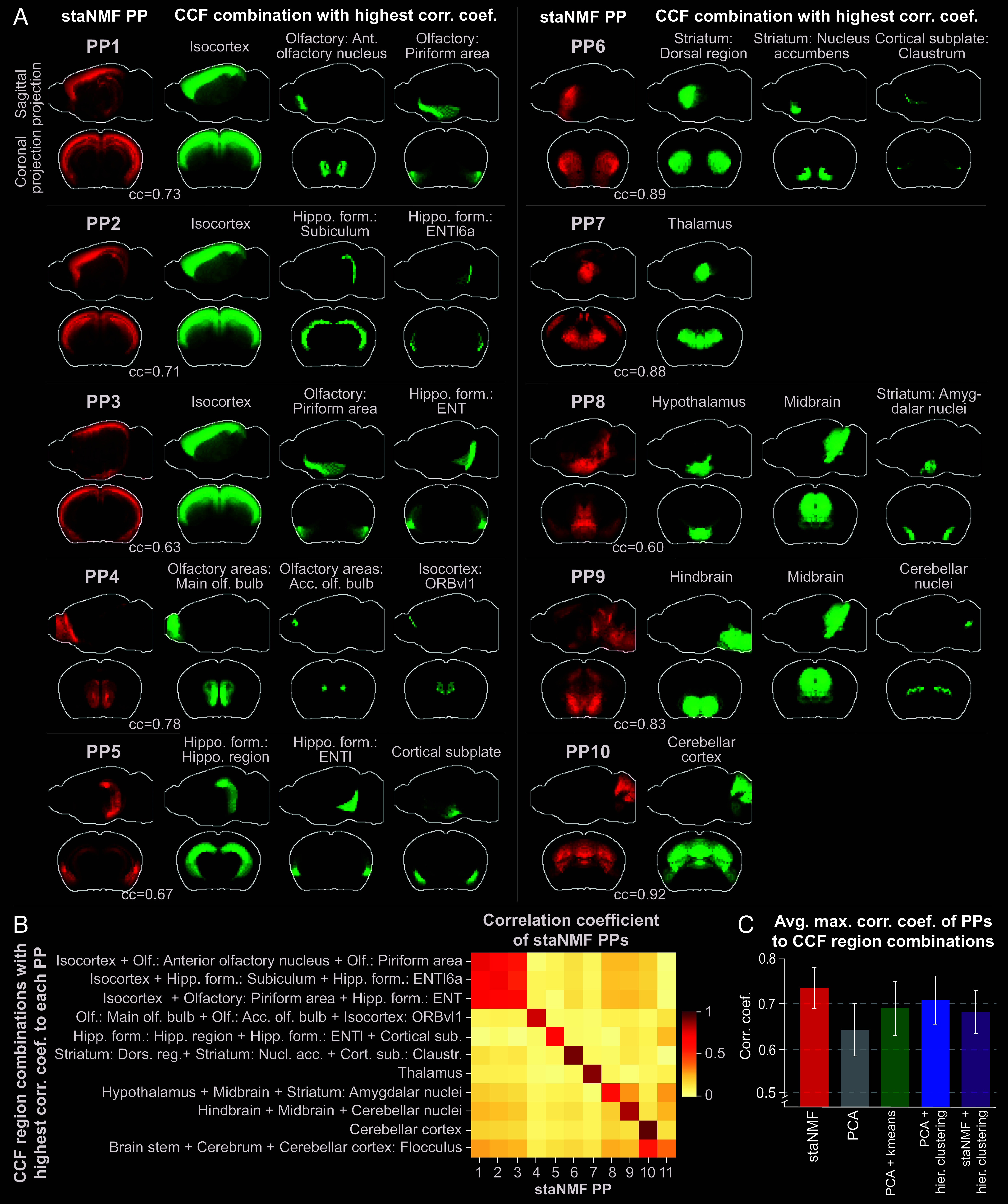
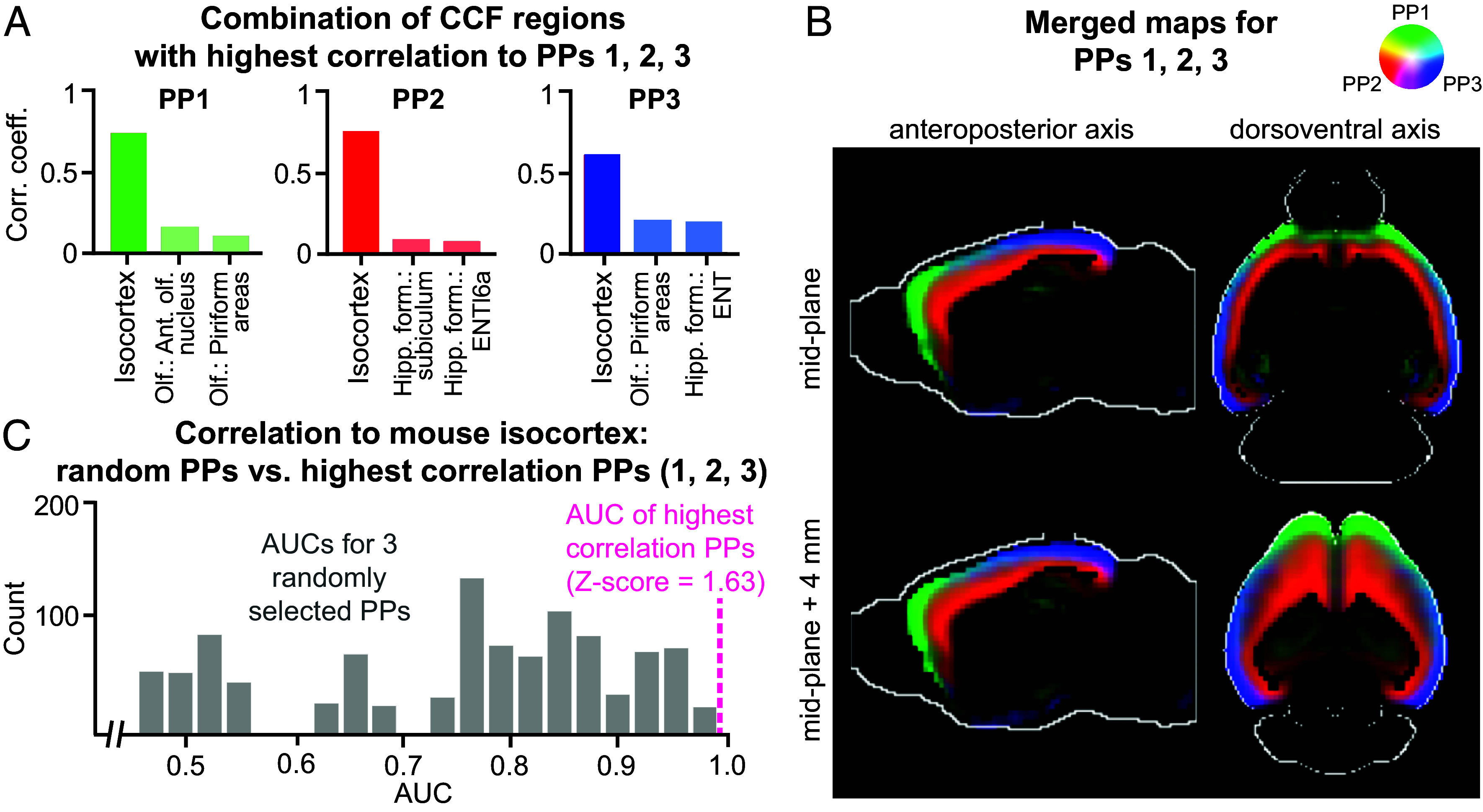
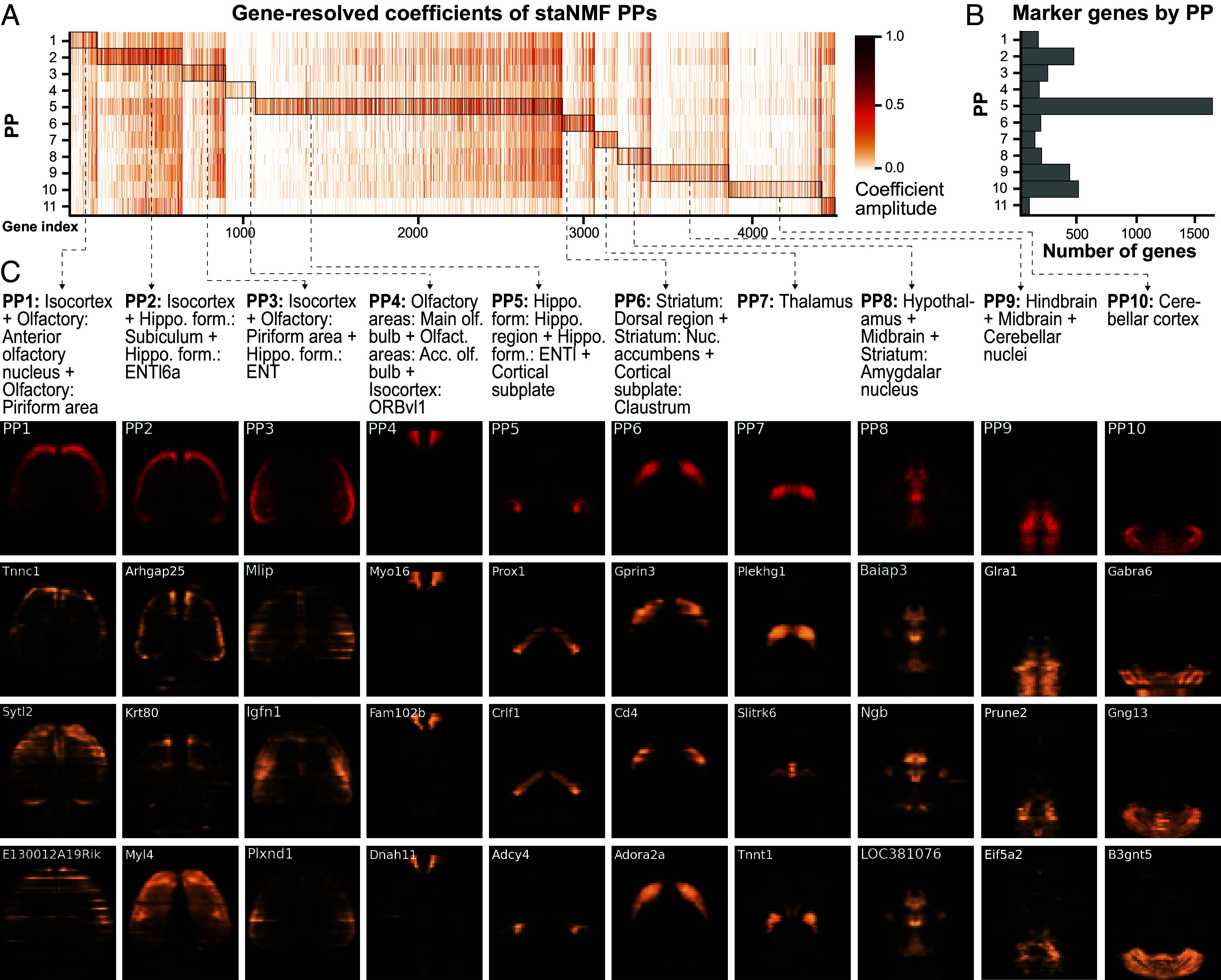
The rapid growth of large-scale spatial gene expression data demands efficient and reliable computational tools to extract major trends of gene expression in their native spatial context. Here, we used stability-driven unsupervised learning (i.e., staNMF) to identify principal patterns (PPs) of 3D gene expression profiles and understand spatial gene distribution and anatomical localization at the whole mouse brain level. Our subsequent spatial correlation analysis systematically compared the PPs to known anatomical regions and ontology from the Allen Mouse Brain Atlas using spatial neighborhoods. We demonstrate that our stable and spatially coherent PPs, whose linear combinations accurately approximate the spatial gene data, are highly correlated with combinations of expert-annotated brain regions. These PPs yield a brain ontology based purely on spatial gene expression. Our PP identification approach outperforms principal component analysis and typical clustering algorithms on the same task. Moreover, we show that the stable PPs reveal marked regional imbalance of brainwide genetic architecture, leading to region-specific marker genes and gene coexpression networks. Our findings highlight the advantages of stability-driven machine learning for plausible biological discovery from dense spatial gene expression data, streamlining tasks that are infeasible by conventional manual approaches.
大规模空间基因表达数据的快速增长需要高效可靠的计算工具,以便在其原生空间背景下提取基因表达的主要趋势。在这里,我们使用稳定性驱动的无监督学习(即 staNMF)来识别 3D 基因表达谱的主要模式 (PP),并了解整个小鼠大脑水平的空间基因分布和解剖定位。我们随后的空间相关分析系统地使用空间邻域将 PPs 与已知的解剖区域和本体从 Allen Mouse Brain Atlas 进行比较。我们证明,我们的稳定且具有空间一致性的 PPs,其线性组合可以准确逼近空间基因数据,与专家注释的脑区组合高度相关。这些 PPs 基于纯粹的空间基因表达产生了一个大脑本体。我们的 PP 识别方法在相同的任务上优于主成分分析和典型的聚类算法。此外,我们表明,稳定的 PPs 揭示了全脑遗传结构的明显区域不平衡,导致特定区域的标记基因和基因共表达网络。我们的研究结果突出了稳定性驱动的机器学习在从密集的空间基因表达数据中进行合理的生物学发现方面的优势,简化了传统手动方法无法完成的任务。