Lopez Romain, Li Baoguo, Keren-Shaul Hadas, Boyeau Pierre, Kedmi Merav, Pilzer David, Jelinski Adam, Yofe Ido, David Eyal, Wagner Allon, Ergen Can, Addadi Yoseph, Golani Ofra, Ronchese Franca, Jordan Michael I, Amit Ido, Yosef Nir
Department of Electrical Engineering and Computer Sciences, University of California, Berkeley, Berkeley CA, USA.
Department of Immunology, Weizmann Institute of Science, Rehovot, Israel.
Nat Biotechnol. 2022 Sep;40(9):1360-1369. doi: 10.1038/s41587-022-01272-8. Epub 2022 Apr 21.
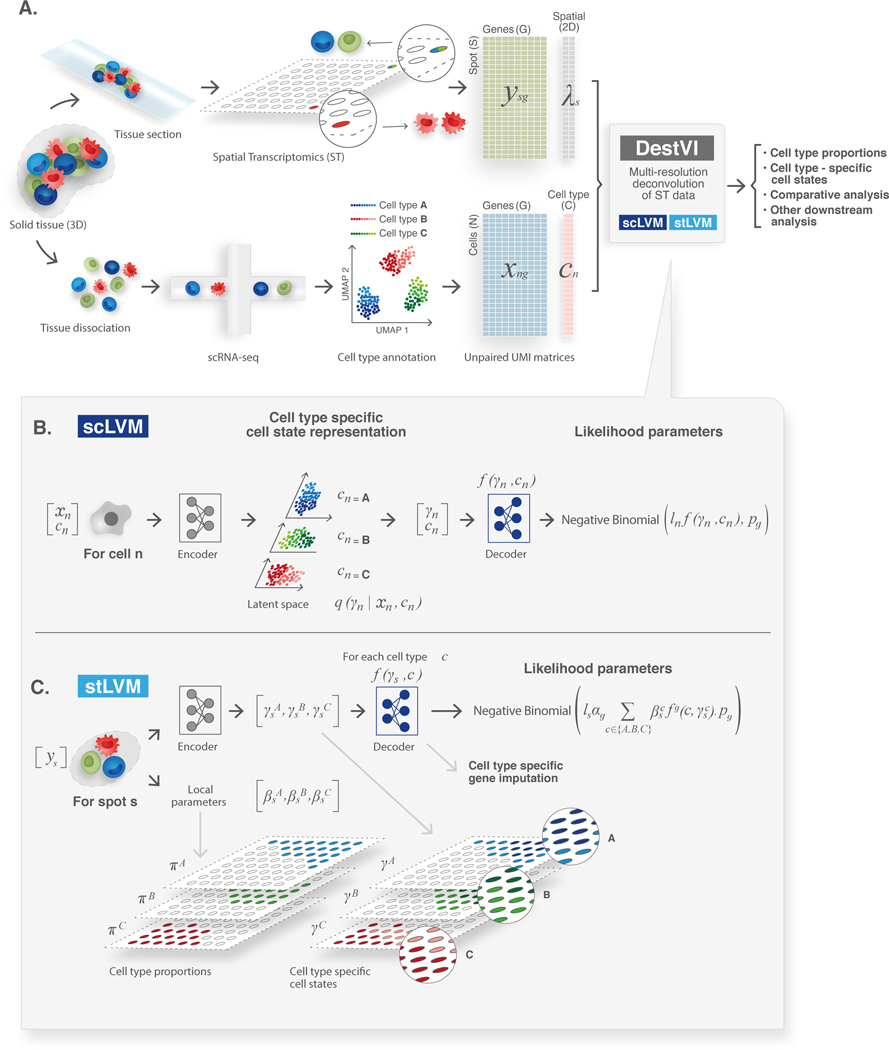
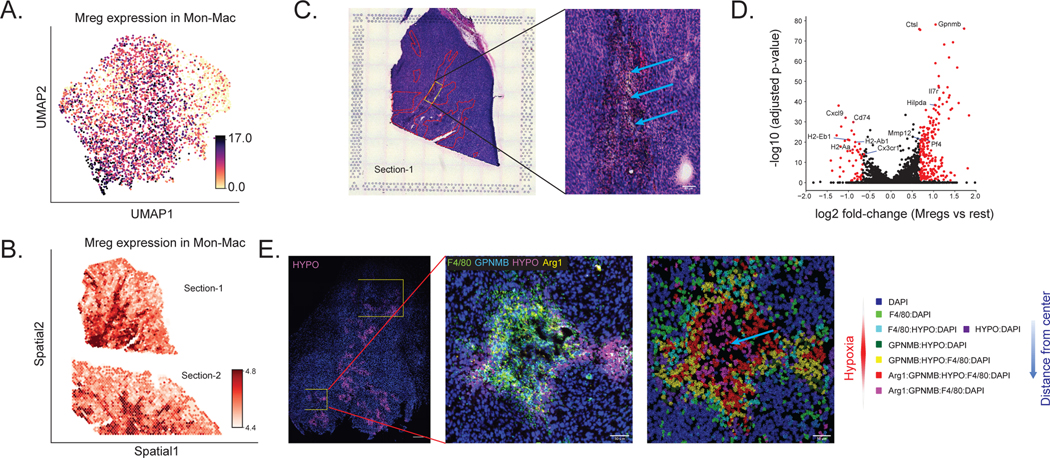
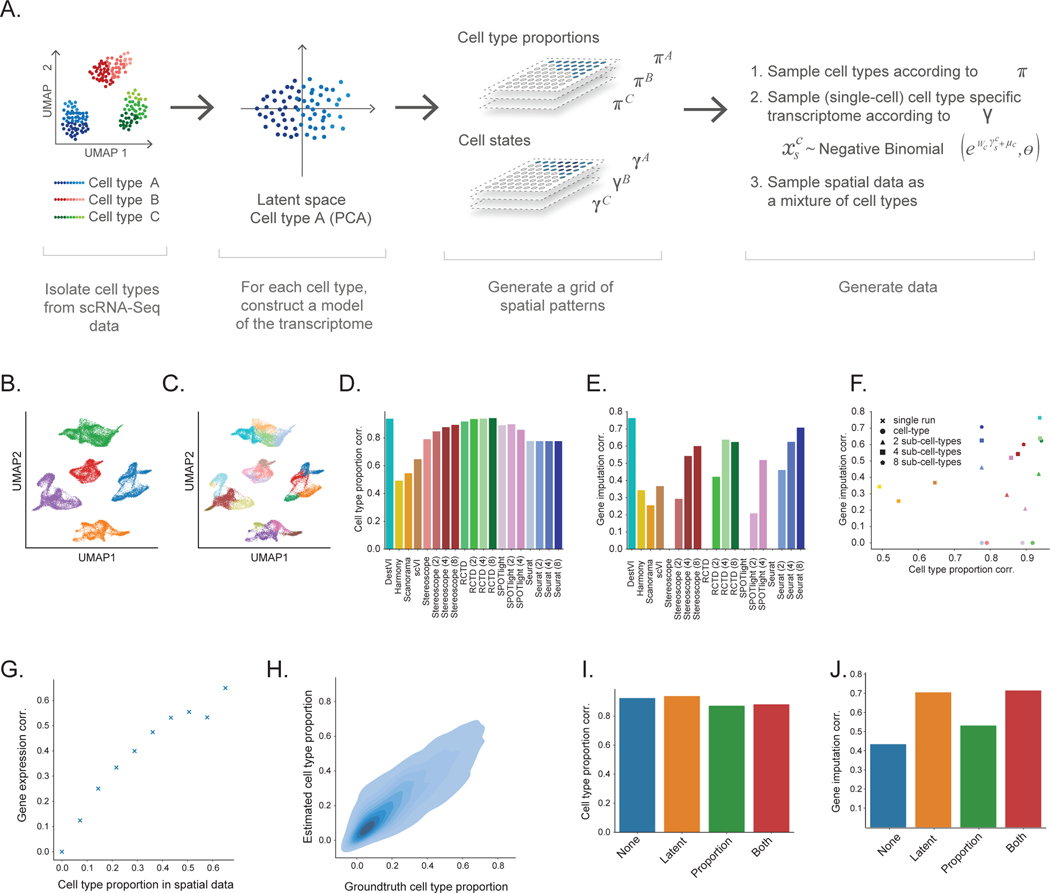
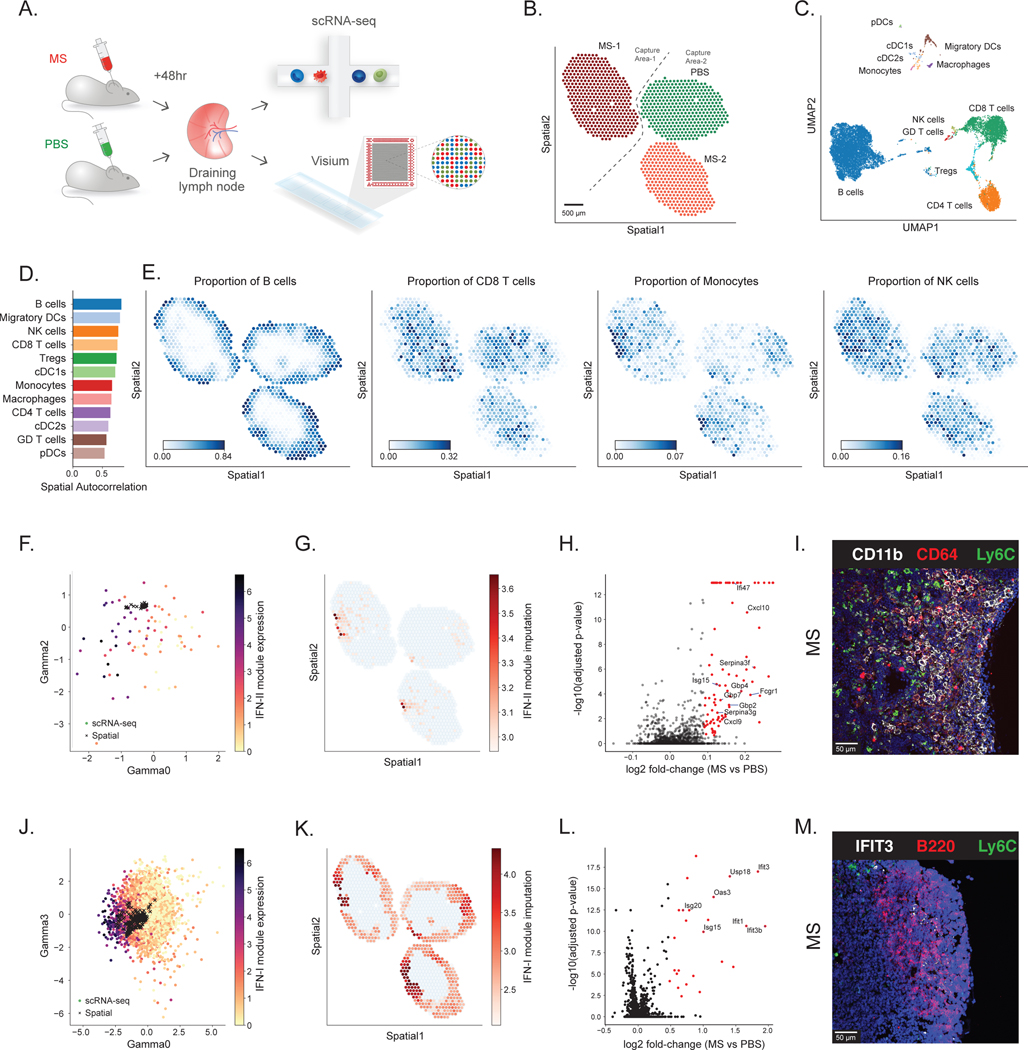
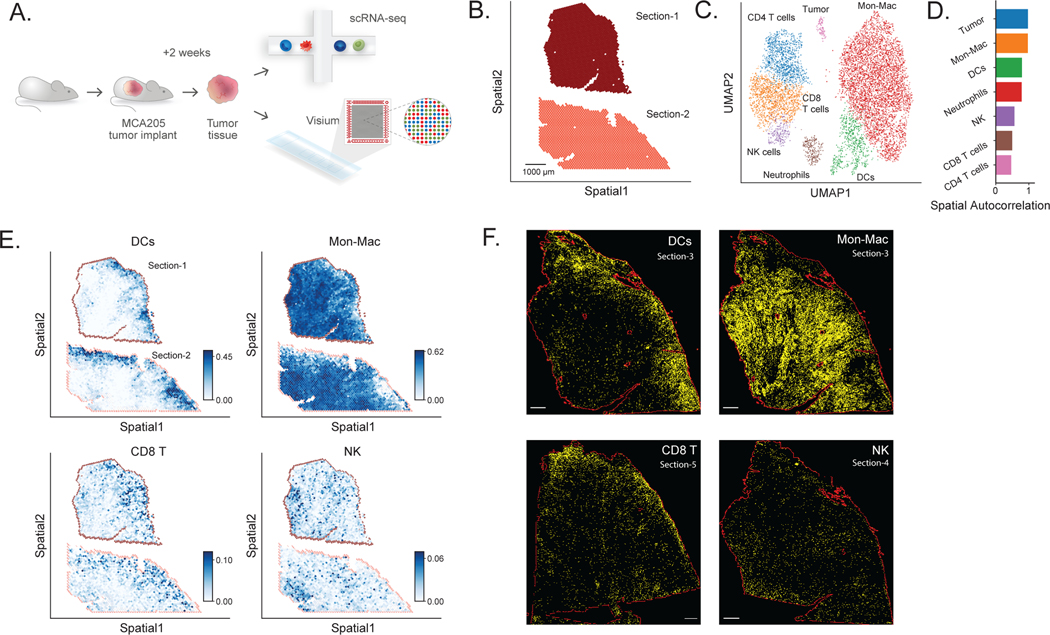
Most spatial transcriptomics technologies are limited by their resolution, with spot sizes larger than that of a single cell. Although joint analysis with single-cell RNA sequencing can alleviate this problem, current methods are limited to assessing discrete cell types, revealing the proportion of cell types inside each spot. To identify continuous variation of the transcriptome within cells of the same type, we developed Deconvolution of Spatial Transcriptomics profiles using Variational Inference (DestVI). Using simulations, we demonstrate that DestVI outperforms existing methods for estimating gene expression for every cell type inside every spot. Applied to a study of infected lymph nodes and of a mouse tumor model, DestVI provides high-resolution, accurate spatial characterization of the cellular organization of these tissues and identifies cell-type-specific changes in gene expression between different tissue regions or between conditions. DestVI is available as part of the open-source software package scvi-tools ( https://scvi-tools.org ).
大多数空间转录组学技术受分辨率限制,其斑点大小大于单个细胞。虽然与单细胞RNA测序的联合分析可以缓解这一问题,但目前的方法仅限于评估离散的细胞类型,揭示每个斑点内细胞类型的比例。为了识别同一类型细胞内转录组的连续变化,我们开发了使用变分推理的空间转录组学图谱反卷积(DestVI)。通过模拟,我们证明DestVI在估计每个斑点内每种细胞类型的基因表达方面优于现有方法。应用于感染淋巴结和小鼠肿瘤模型的研究中,DestVI提供了这些组织细胞组织的高分辨率、准确的空间特征,并识别了不同组织区域之间或不同条件之间基因表达的细胞类型特异性变化。DestVI作为开源软件包scvi-tools(https://scvi-tools.org)的一部分可用。