Université Paris-Saclay, Gustave Roussy, Inserm U981, Villejuif, France.
Département d'Innovation Thérapeutique (DITEP), Gustave Roussy, Villejuif, France.
Clin Cancer Res. 2024 Nov 1;30(21):4943-4956. doi: 10.1158/1078-0432.CCR-24-1834.
Understanding resistance to selective FGFR inhibitors is crucial to improve the clinical outcomes of patients with FGFR2-driven malignancies.
We analyzed sequential ctDNA, ± whole-exome sequencing, or targeted next-generation sequencing on tissue biopsies from patients with tumors harboring activating FGFR2 alterations progressing on pan-FGFR-selective inhibitors, collected in the prospective UNLOCK program. FGFR2::BICC1 Ba/F3 and patient-derived xenograft models were used for functional studies.
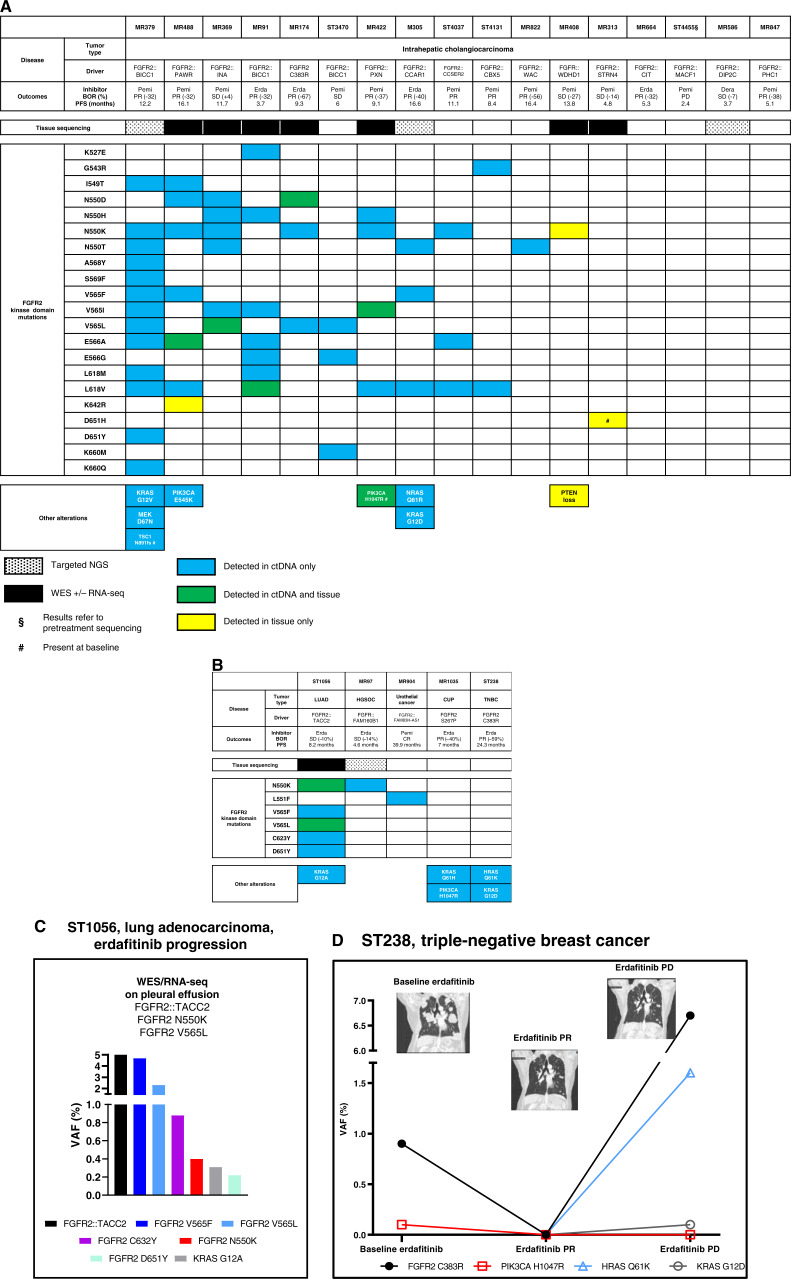
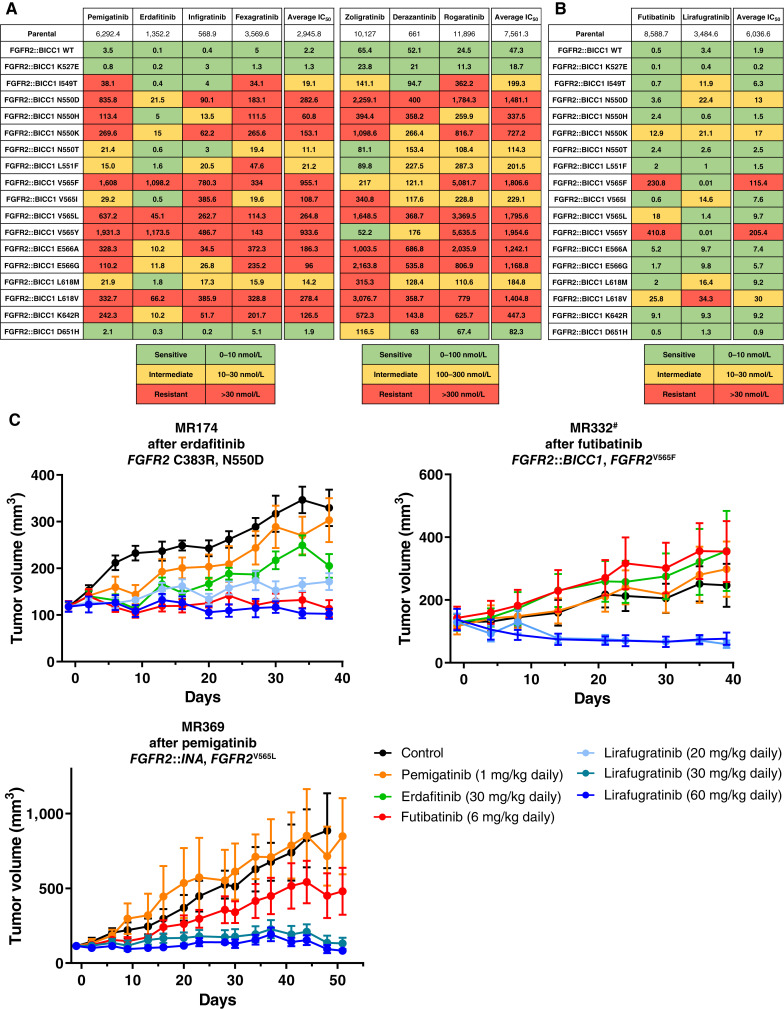
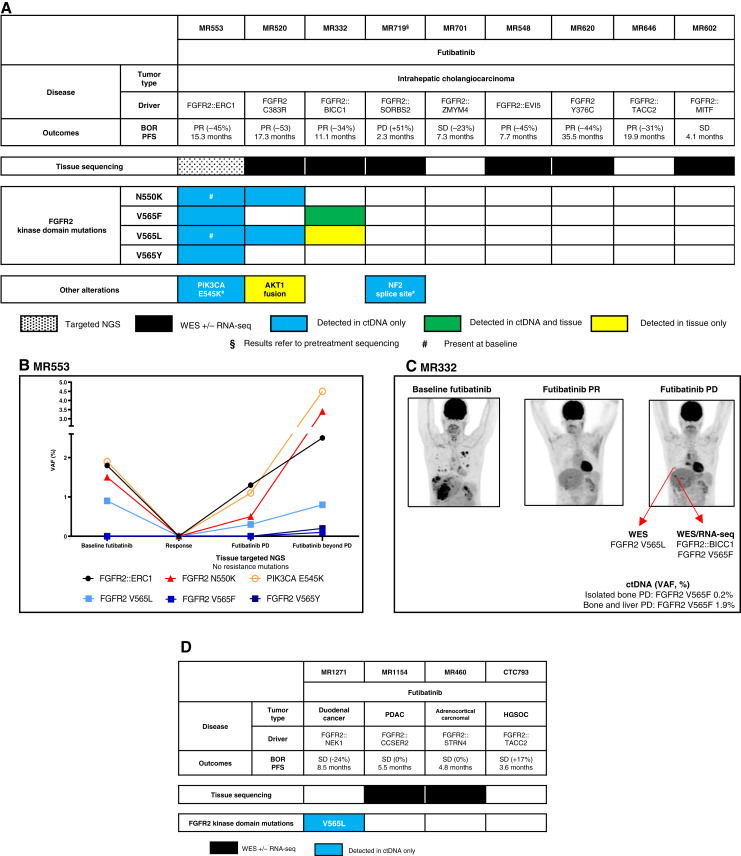
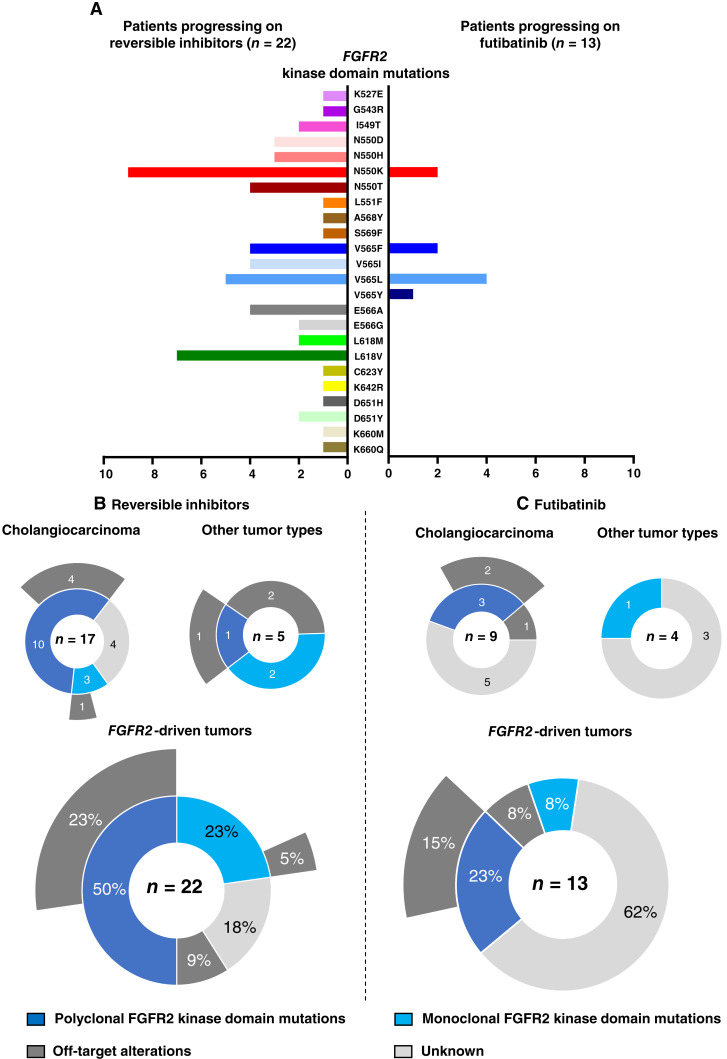
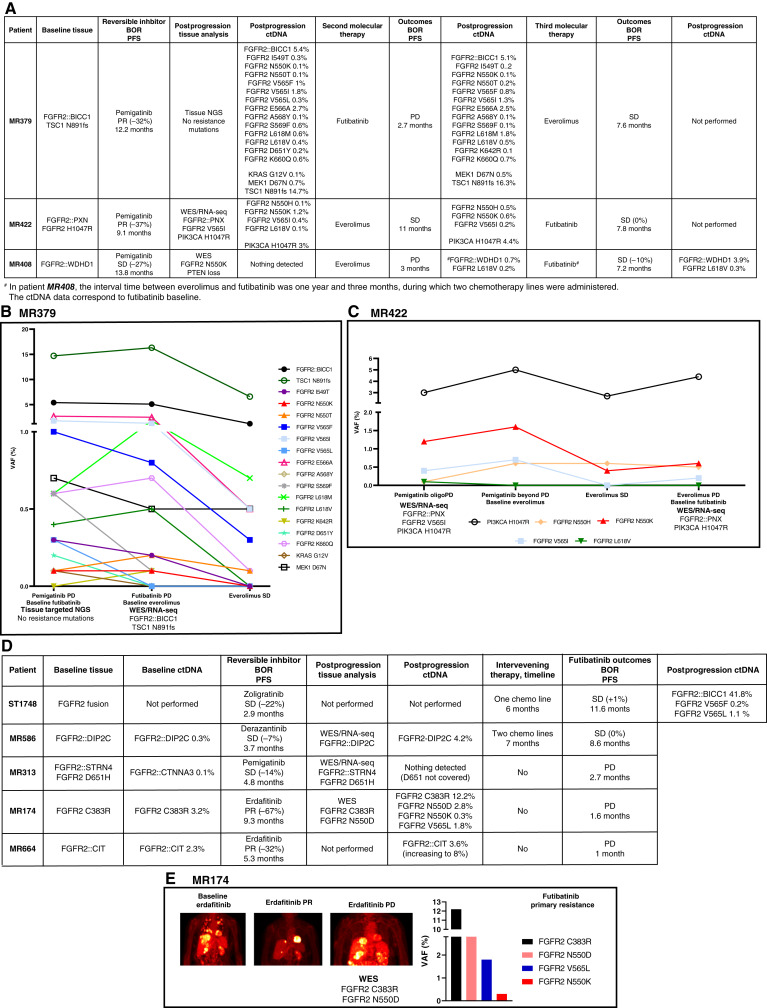
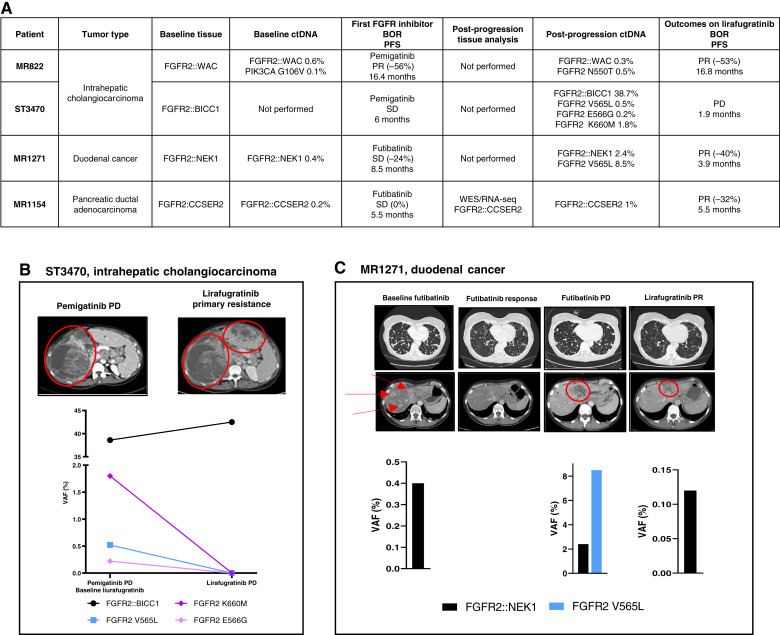
Thirty-six patients were included. In cholangiocarcinoma, at resistance to both reversible inhibitors (e.g., pemigatinib and erdafitinib) and the irreversible inhibitor futibatinib, polyclonal FGFR2 kinase domain mutations were frequent (14/27 patients). Tumors other than cholangiocarcinoma shared the same mutated FGFR2 residues, but polyclonality was rare (1/9 patients). At resistance to reversible inhibitors, 14 residues in the FGFR2 kinase domain were mutated-after futibatinib, only the molecular brake N550 and the gatekeeper V565. Off-target alterations in PI3K/mTOR and MAPK pathways were found in 11 patients, often together with on-target mutations. At progression to a first FGFR inhibitor, 12 patients received futibatinib or lirafugratinib (irreversible inhibitors), with variable clinical outcomes depending on previous resistance mechanisms. Two patients with TSC1 or PIK3CA mutations benefited from everolimus. In cell viability assays on Ba/F3 and in pharmacologic studies on patient-derived xenografts, irreversible inhibitors retained better activity against FGFR2 kinase domain mutations, with lirafugratinib active against the recalcitrant V565L/F/Y.
At progression to FGFR inhibitors, FGFR2-driven malignancies are characterized by high intra- and interpatient molecular heterogeneity, particularly in cholangiocarcinoma. Resistance to FGFR inhibitors can be overcome by sequential, molecularly oriented treatment strategies across FGFR2-driven tumors.
了解对选择性 FGFR 抑制剂的耐药性对于改善 FGFR 驱动的恶性肿瘤患者的临床结局至关重要。
我们分析了在 UNLOCK 前瞻性研究中收集的,携带激活 FGFR2 改变的肿瘤患者在接受泛 FGFR 选择性抑制剂治疗进展后的连续 ctDNA、± 全外显子组测序或靶向下一代测序的组织活检。使用 FGFR2::BICC1 Ba/F3 和患者来源的异种移植模型进行功能研究。
共纳入 36 例患者。在胆管癌中,对于两种可逆抑制剂(如培米替尼和厄达替尼)和不可逆抑制剂富替替尼耐药时,FGFR2 激酶结构域的多克隆突变很常见(27 例患者中有 14 例)。胆管癌以外的肿瘤共享相同的突变 FGFR2 残基,但很少有多克隆性(9 例患者中有 1 例)。对于可逆抑制剂耐药时,FGFR2 激酶结构域中有 14 个残基发生突变——在富替替尼耐药后,只有分子刹车 N550 和门控 V565 发生突变。在 11 例患者中发现了 PI3K/mTOR 和 MAPK 通路的非靶点改变,通常与靶点突变一起存在。在进展为第一代 FGFR 抑制剂时,12 例患者接受了富替替尼或来伐替尼(不可逆抑制剂)治疗,根据先前的耐药机制,临床结局各不相同。2 例 TSC1 或 PIK3CA 突变的患者受益于依维莫司。在 Ba/F3 的细胞活力测定和患者来源的异种移植的药物研究中,不可逆抑制剂对 FGFR2 激酶结构域突变保留了更好的活性,来伐替尼对难治性 V565L/F/Y 有效。
在进展为 FGFR 抑制剂时,FGFR 驱动的恶性肿瘤表现出高度的肿瘤内和肿瘤间分子异质性,特别是在胆管癌中。通过跨 FGFR2 驱动肿瘤的顺序、分子靶向治疗策略可以克服 FGFR 抑制剂的耐药性。