Weinstock Joshua S, Chaudhry Sharjeel A, Ioannou Maria, Viskadourou Maria, Reventun Paula, Jakubek Yasminka A, Liggett L Alexander, Laurie Cecelia, Broome Jai G, Khan Alyna, Taylor Kent D, Guo Xiuqing, Peyser Patricia A, Boerwinkle Eric, Chami Nathalie, Kenny Eimear E, Loos Ruth J, Psaty Bruce M, Russell Tracy P, Brody Jennifer A, Yun Jeong H, Cho Michael H, Vasan Ramachandran S, Kardia Sharon L, Smith Jennifer A, Raffield Laura M, Bidulescu Aurelian, O'Brien Emily, de Andrade Mariza, Rotter Jerome I, Rich Stephen S, Tracy Russell P, Chen Yii Der Ida, Gu C Charles, Hsiung Chao A, Kooperberg Charles, Haring Bernhard, Nassir Rami, Mathias Rasika, Reiner Alex, Sankaran Vijay, Lowenstein Charles J, Blackwell Thomas W, Abecasis Goncalo R, Smith Albert V, Kang Hyun M, Natarajan Pradeep, Jaiswal Siddhartha, Bick Alexander, Post Wendy S, Scheet Paul, Auer Paul, Karantanos Theodoros, Battle Alexis, Arvanitis Marios
Department of Human Genetics, School of Medicine, Emory University, Atlanta, GA, USA.
Division of Cardiology, Department of Medicine, Johns Hopkins University, Baltimore, MD.
medRxiv. 2024 Aug 26:2024.08.22.24312319. doi: 10.1101/2024.08.22.24312319.
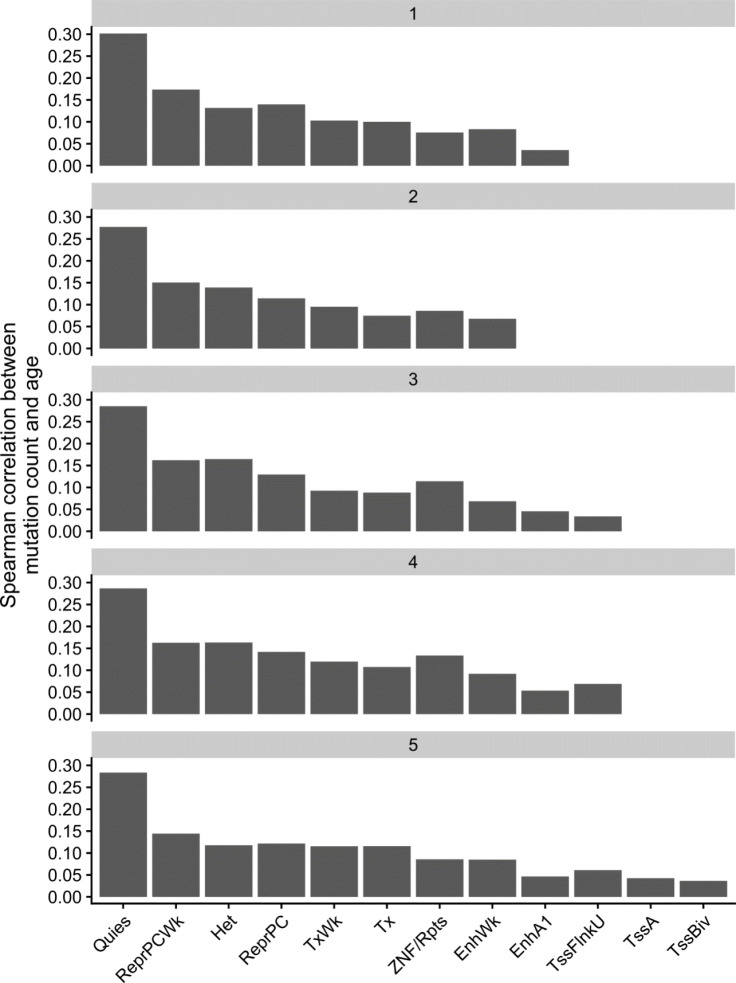
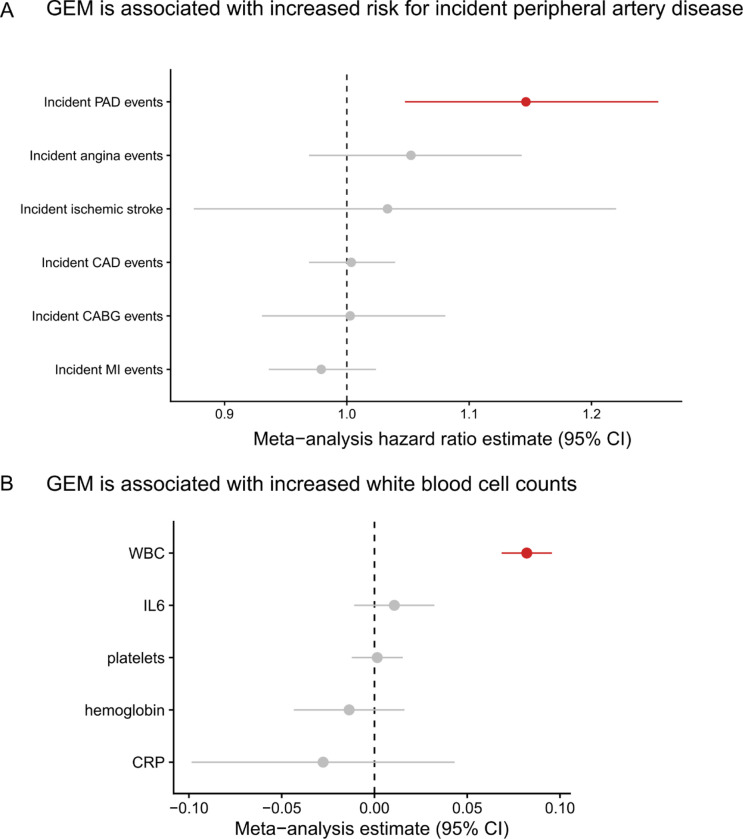
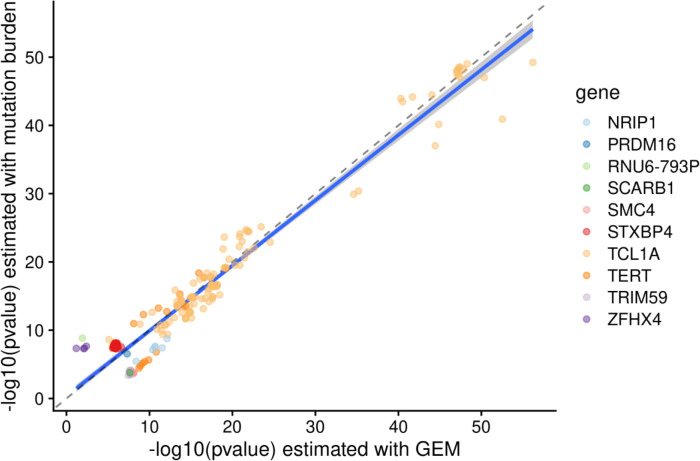
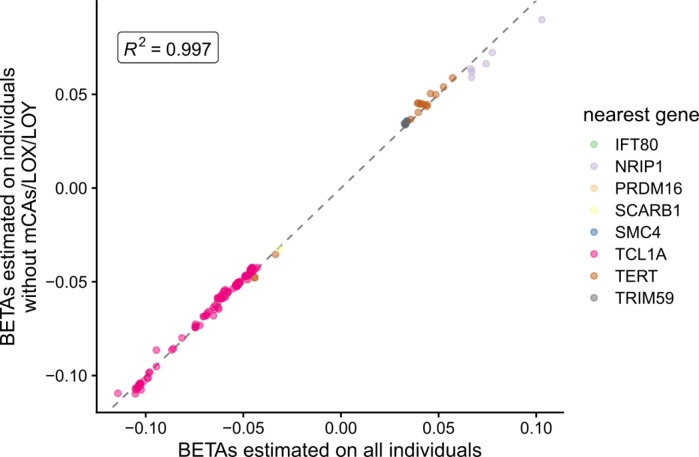
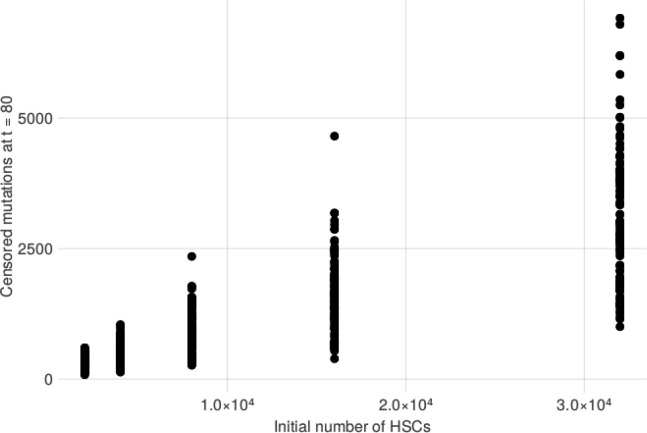
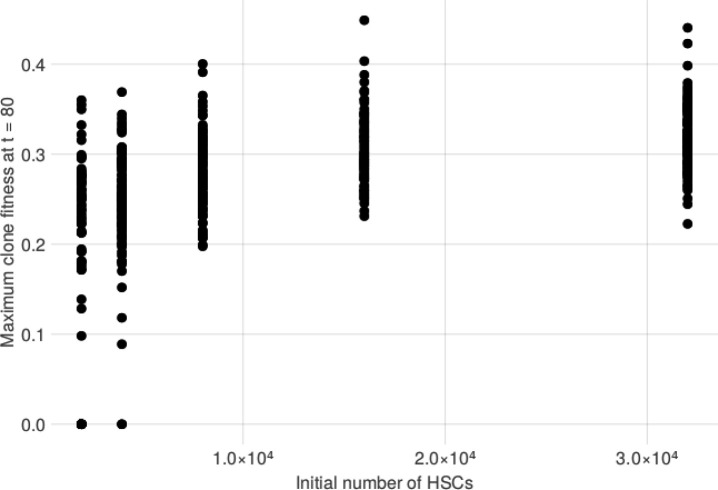
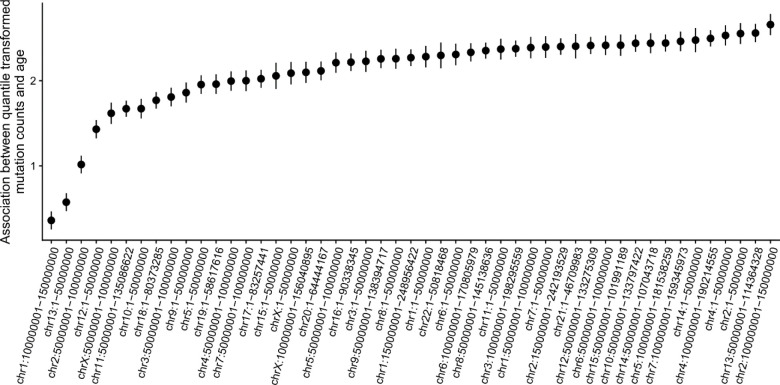
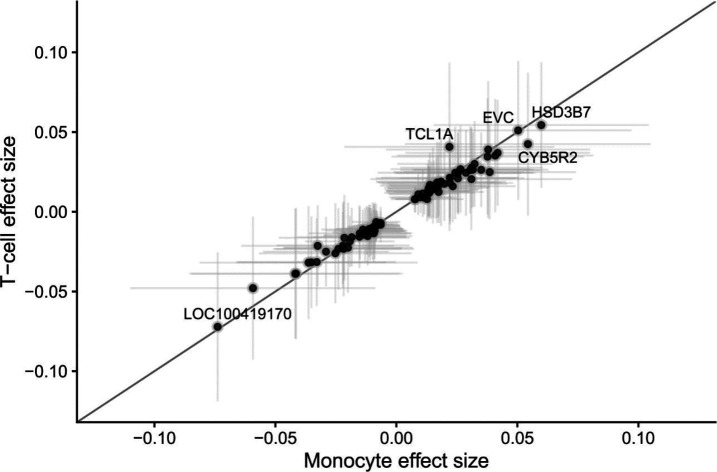
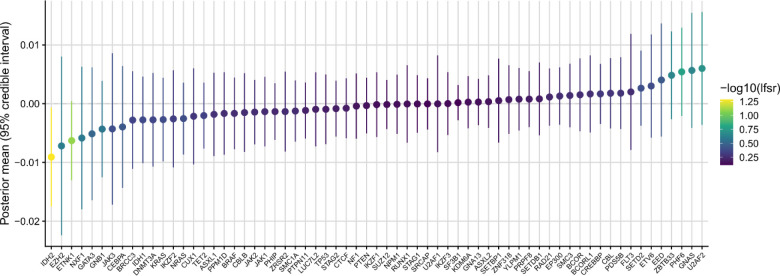
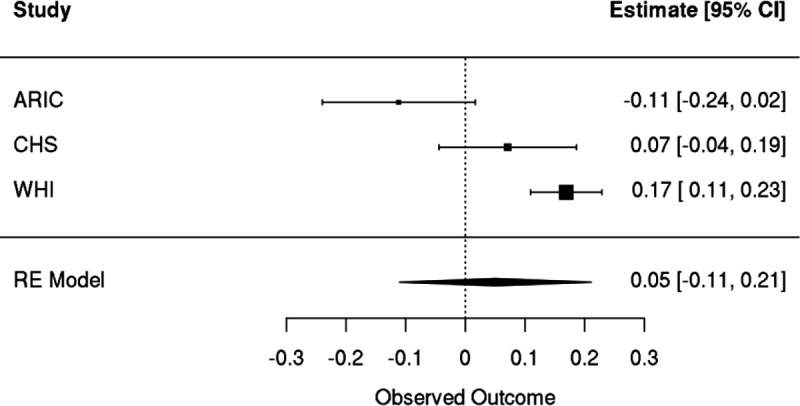
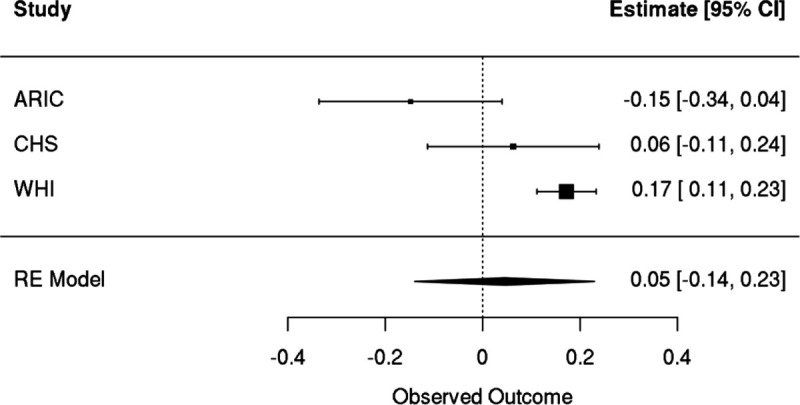
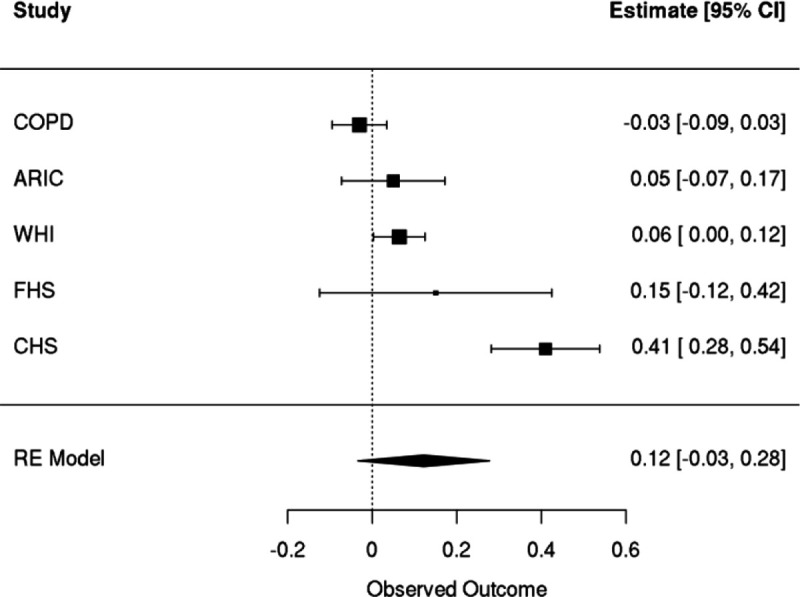
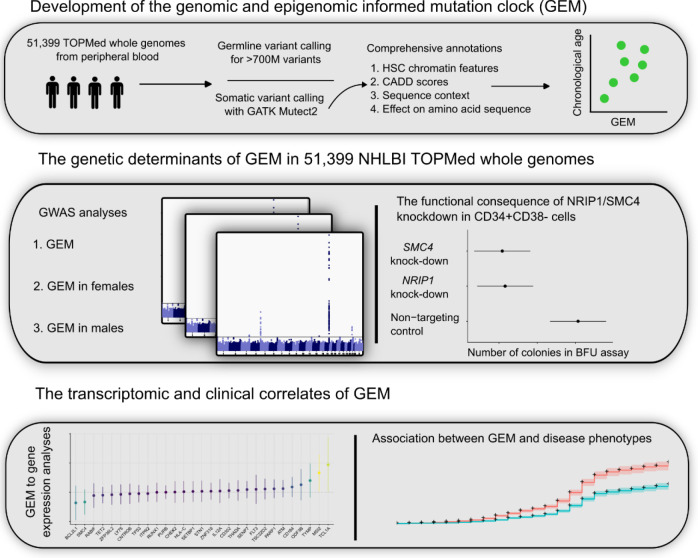
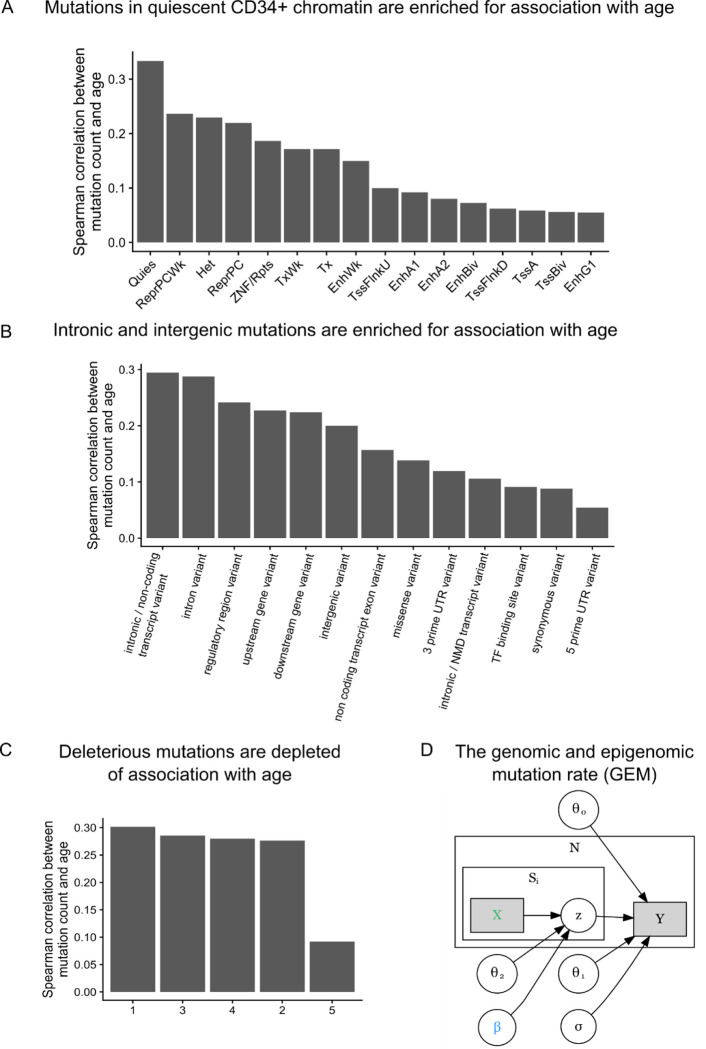
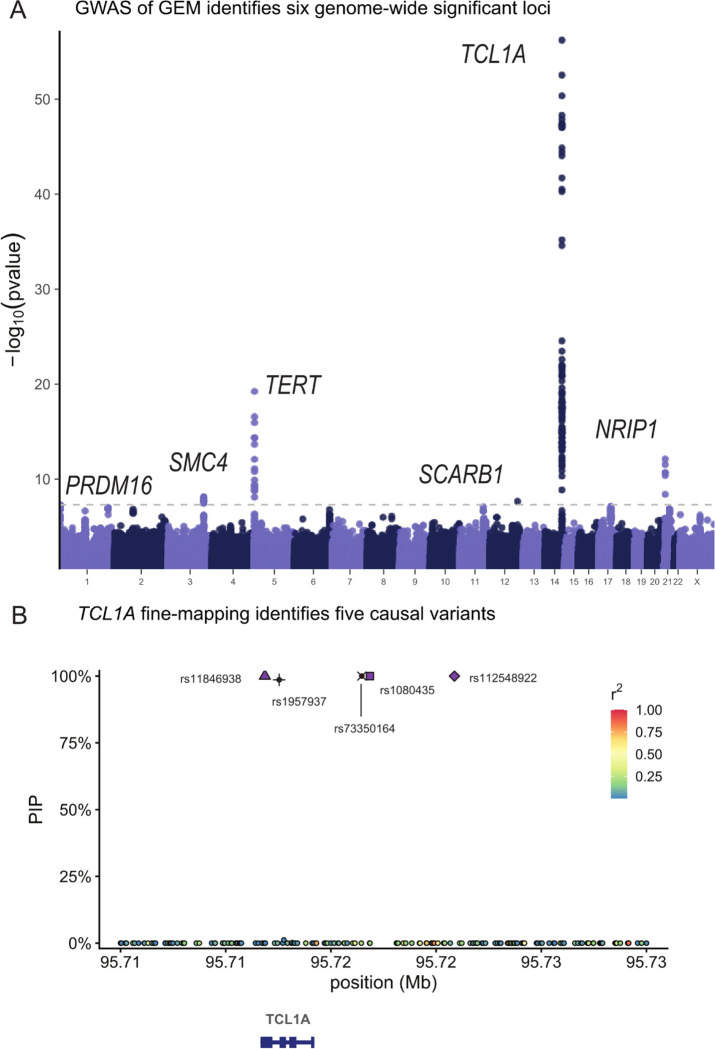
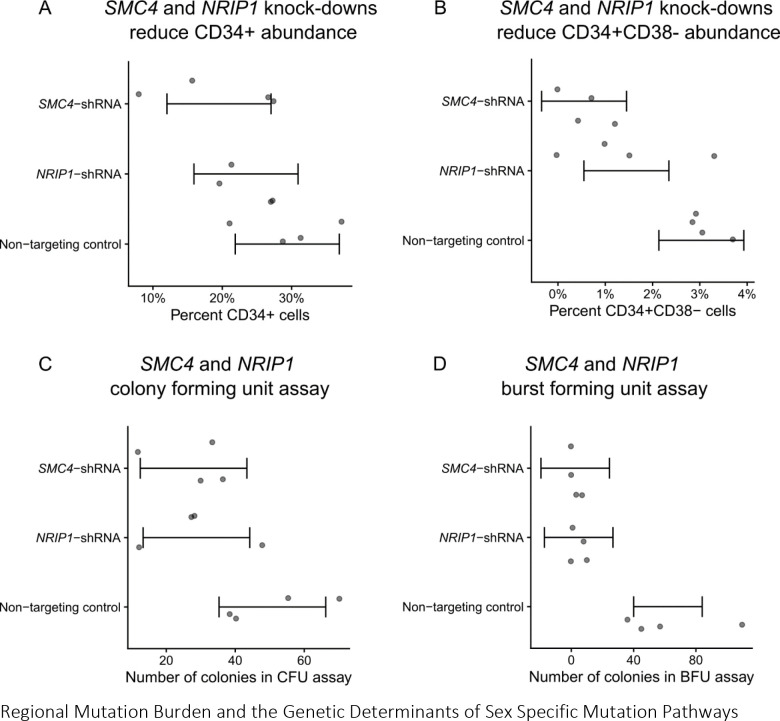
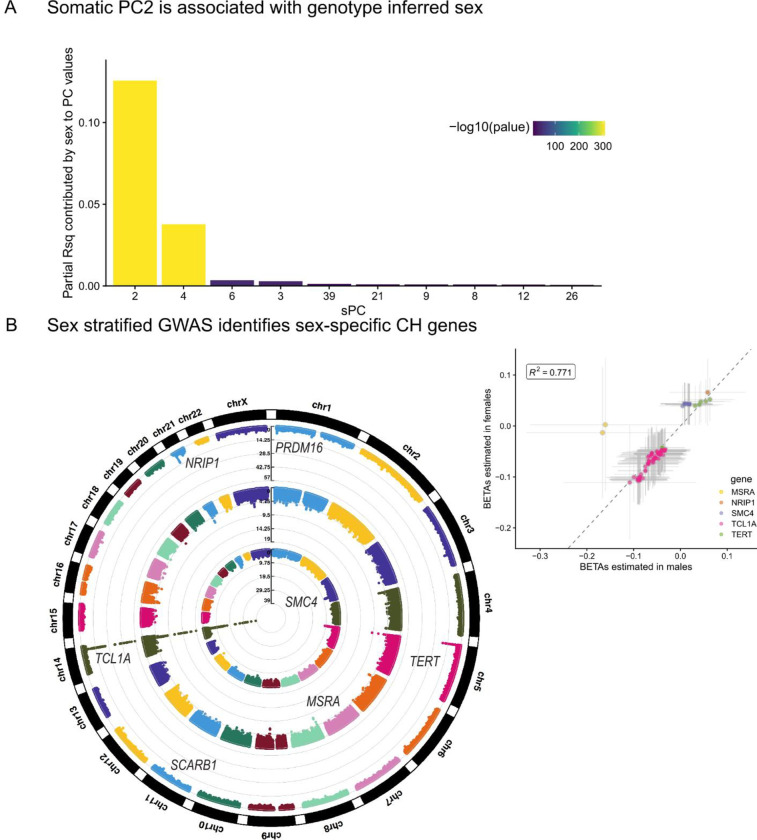
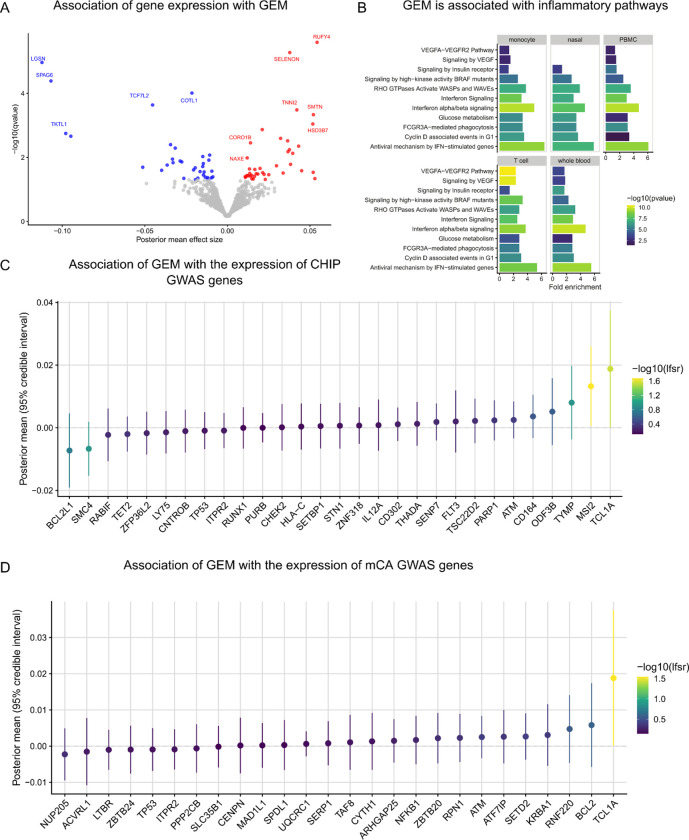
Clonal hematopoiesis (CH) is defined by the expansion of a lineage of genetically identical cells in blood. Genetic lesions that confer a fitness advantage, such as point mutations or mosaic chromosomal alterations (mCAs) in genes associated with hematologic malignancy, are frequent mediators of CH. However, recent analyses of both single cell-derived colonies of hematopoietic cells and population sequencing cohorts have revealed CH frequently occurs in the absence of known driver genetic lesions. To characterize CH without known driver genetic lesions, we used 51,399 deeply sequenced whole genomes from the NHLBI TOPMed sequencing initiative to perform simultaneous germline and somatic mutation analyses among individuals without leukemogenic point mutations (LPM), which we term CH-LPMneg. We quantified CH by estimating the total mutation burden. Because estimating somatic mutation burden without a paired-tissue sample is challenging, we developed a novel statistical method, the Genomic and Epigenomic informed Mutation (GEM) rate, that uses external genomic and epigenomic data sources to distinguish artifactual signals from true somatic mutations. We performed a genome-wide association study of GEM to discover the germline determinants of CH-LPMneg. After fine-mapping and variant-to-gene analyses, we identified seven genes associated with CH-LPMneg (, , ), and one locus associated with a sex-associated mutation pathway (. We performed a secondary analysis excluding individuals with mCAs, finding that the genetic architecture was largely unaffected by their inclusion. Functional analyses of and implicated altered HSC self-renewal and proliferation as the primary mediator of mutation burden in blood. We then performed comprehensive multi-tissue transcriptomic analyses, finding that the expression levels of 404 genes are associated with GEM. Finally, we performed phenotypic association meta-analyses across four cohorts, finding that GEM is associated with increased white blood cell count and increased risk for incident peripheral artery disease, but is not significantly associated with incident stroke or coronary disease events. Overall, we develop GEM for quantifying mutation burden from WGS without a paired-tissue sample and use GEM to discover the genetic, genomic, and phenotypic correlates of CH-LPMneg.
克隆性造血(CH)定义为血液中基因相同的细胞系的扩增。赋予适应性优势的基因损伤,如与血液系统恶性肿瘤相关基因中的点突变或嵌合染色体改变(mCAs),是CH常见的介导因素。然而,最近对造血细胞单细胞衍生集落和群体测序队列的分析表明,CH经常在没有已知驱动基因损伤的情况下发生。为了表征没有已知驱动基因损伤的CH,我们使用了来自美国国立卫生研究院心肺血液研究所(NHLBI)TOPMed测序计划的51399个深度测序的全基因组,对没有致白血病点突变(LPM)的个体进行种系和体细胞突变同步分析,我们将其称为CH-LPMneg。我们通过估计总突变负担来量化CH。由于在没有配对组织样本的情况下估计体细胞突变负担具有挑战性,我们开发了一种新的统计方法,即基因组和表观基因组信息突变(GEM)率,该方法使用外部基因组和表观基因组数据源来区分人为信号和真正的体细胞突变。我们对GEM进行了全基因组关联研究,以发现CH-LPMneg的种系决定因素。经过精细定位和变异到基因分析,我们确定了7个与CH-LPMneg相关的基因(,,),以及1个与性别相关突变途径相关的位点(。我们进行了一项排除mCAs个体的二次分析,发现基因结构在很大程度上不受其纳入的影响。对和的功能分析表明,造血干细胞自我更新和增殖的改变是血液中突变负担的主要介导因素。然后,我们进行了全面的多组织转录组分析,发现404个基因的表达水平与GEM相关。最后,我们对四个队列进行了表型关联荟萃分析,发现GEM与白细胞计数增加和外周动脉疾病发病风险增加相关,但与中风或冠心病事件的发生无显著关联。总体而言,我们开发了GEM用于在没有配对组织样本的情况下从全基因组测序中量化突变负担,并使用GEM发现CH-LPMneg的遗传、基因组和表型相关性。