Williams Marc J, Vázquez-García Ignacio, Tam Grittney, Wu Michelle, Varice Nancy, Havasov Eliyahu, Shi Hongyu, Satas Gryte, Lees Hannah J, Lee Jake June-Koo, Myers Matthew A, Zatzman Matthew, Rusk Nicole, Ali Emily, Shah Ronak H, Berger Michael F, Mohibullah Neeman, Lakhman Yulia, Chi Dennis S, Abu-Rustum Nadeem R, Aghajanian Carol, McPherson Andrew, Zamarin Dmitriy, Loomis Brian, Weigelt Britta, Friedman Claire F, Shah Sohrab P
Computational Oncology, Department of Epidemiology and Biostatistics, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA.
The Halvorsen Center for Computational Oncology, Memorial Sloan Kettering Cancer Center, New York, NY, USA.
bioRxiv. 2024 Aug 23:2024.08.21.609031. doi: 10.1101/2024.08.21.609031.
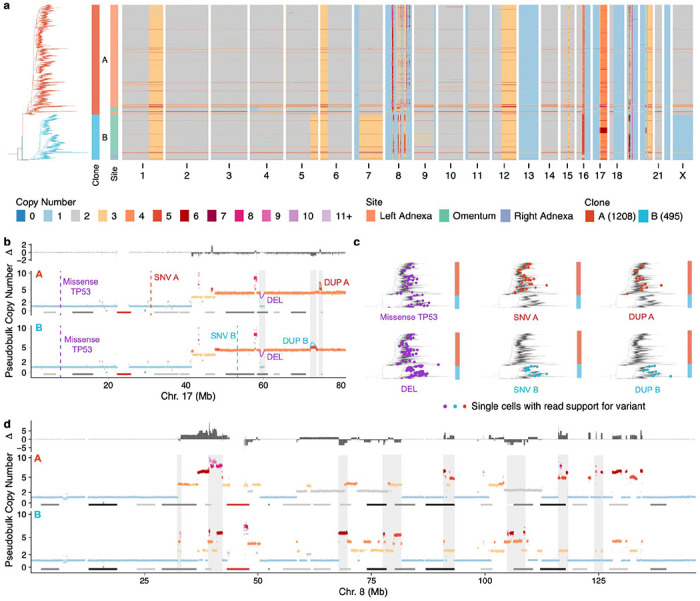
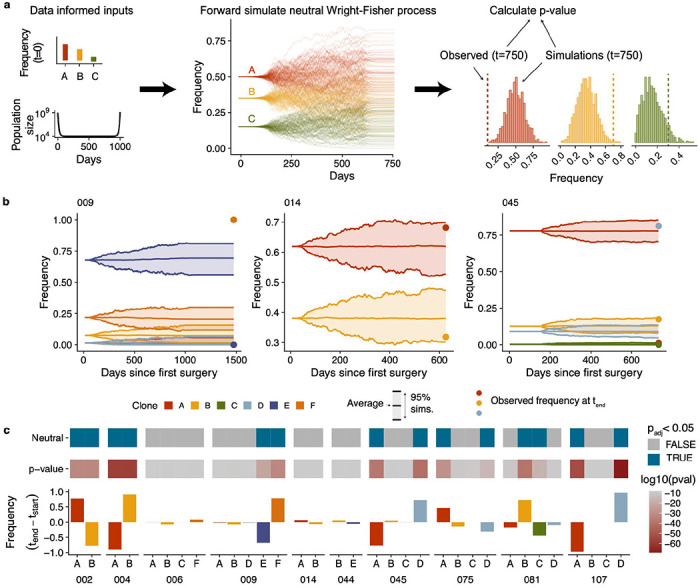
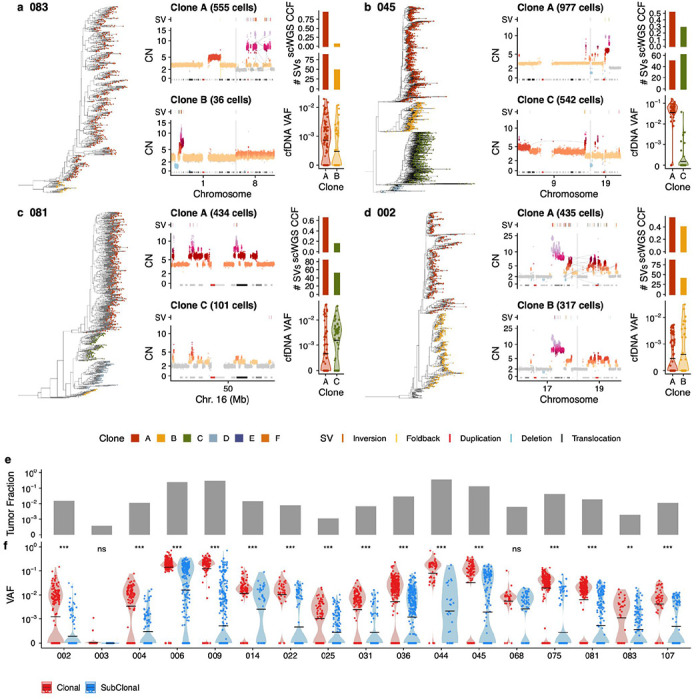
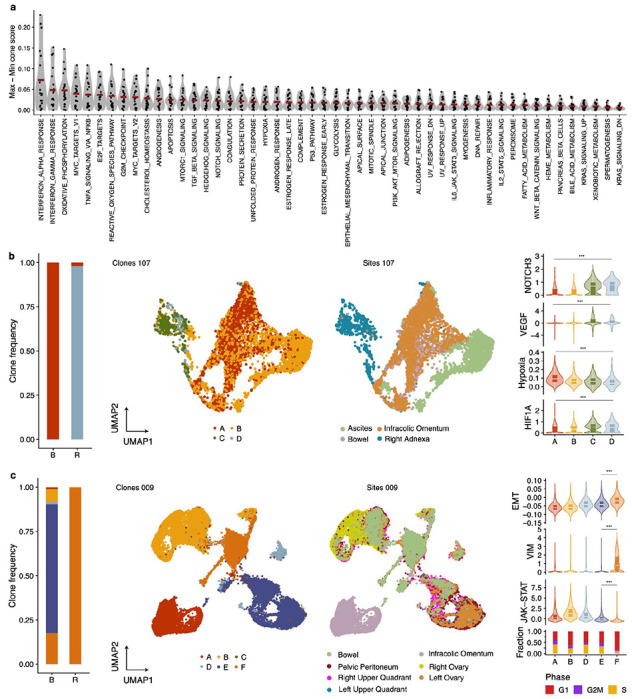
Drug resistance is the major cause of therapeutic failure in high-grade serous ovarian cancer (HGSOC). Yet, the mechanisms by which tumors evolve to drug resistant states remains largely unknown. To address this, we aimed to exploit clone-specific genomic structural variations by combining scaled single-cell whole genome sequencing with longitudinally collected cell-free DNA (cfDNA), enabling clonal tracking before, during and after treatment. We developed a cfDNA hybrid capture, deep sequencing approach based on leveraging clone-specific structural variants as endogenous barcodes, with orders of magnitude lower error rates than single nucleotide variants in ctDNA (circulating tumor DNA) detection, demonstrated on 19 patients at baseline. We then applied this to monitor and model clonal evolution over several years in ten HGSOC patients treated with systemic therapy from diagnosis through recurrence. We found drug resistance to be polyclonal in most cases, but frequently dominated by a single high-fitness and expanding clone, reducing clonal diversity in the relapsed disease state in most patients. Drug-resistant clones frequently displayed notable genomic features, including high-level amplifications of oncogenes such as , , , and . Using a population genetics Wright-Fisher model, we found evolutionary trajectories of these features were consistent with drug-induced positive selection. In select cases, these alterations impacted selection of secondary lines of therapy with positive patient outcomes. For cases with matched single-cell RNA sequencing data, pre-existing and genomically encoded phenotypic states such as upregulation of EMT and VEGF were linked to drug resistance. Together, our findings indicate that drug resistant states in HGSOC pre-exist at diagnosis and lead to dramatic clonal expansions that alter clonal composition at the time of relapse. We suggest that combining tumor single cell sequencing with cfDNA enables clonal tracking in patients and harbors potential for evolution-informed adaptive treatment decisions.
耐药性是高级别浆液性卵巢癌(HGSOC)治疗失败的主要原因。然而,肿瘤演变为耐药状态的机制在很大程度上仍不清楚。为了解决这个问题,我们旨在通过将规模化单细胞全基因组测序与纵向收集的游离DNA(cfDNA)相结合,利用克隆特异性基因组结构变异,从而能够在治疗前、治疗期间和治疗后进行克隆追踪。我们基于利用克隆特异性结构变异作为内源性条形码,开发了一种cfDNA杂交捕获、深度测序方法,在ctDNA(循环肿瘤DNA)检测中,其错误率比单核苷酸变异低几个数量级,在19例患者的基线期得到了验证。然后,我们将此方法应用于监测和模拟10例接受全身治疗的HGSOC患者从诊断到复发数年的克隆进化情况。我们发现,在大多数情况下,耐药是多克隆性的,但通常由单个高适应性且不断扩增的克隆主导,这导致大多数患者复发疾病状态下的克隆多样性降低。耐药克隆经常表现出显著的基因组特征,包括某些癌基因如、、、和的高水平扩增。使用群体遗传学的赖特 - 费希尔模型,我们发现这些特征的进化轨迹与药物诱导的正选择一致。在某些情况下,这些改变影响了二线治疗方案的选择,并取得了积极的患者治疗效果。对于具有匹配单细胞RNA测序数据的病例,预先存在的以及基因组编码的表型状态,如EMT和VEGF的上调与耐药性相关。总之,我们的研究结果表明,HGSOC中的耐药状态在诊断时就已存在,并导致显著的克隆扩增,从而在复发时改变克隆组成。我们建议,将肿瘤单细胞测序与cfDNA相结合能够对患者进行克隆追踪,并具有为基于进化信息的适应性治疗决策提供依据的潜力。