Eye Research Institute, Oakland University, Rochester, MI 48309, United States.
Stein Eye Institute, David Geffen School of Medicine, University of California Los Angeles, Los Angeles, CA 90095-7000, United States.
Hum Mol Genet. 2024 Nov 5;33(21):1916-1928. doi: 10.1093/hmg/ddae128.
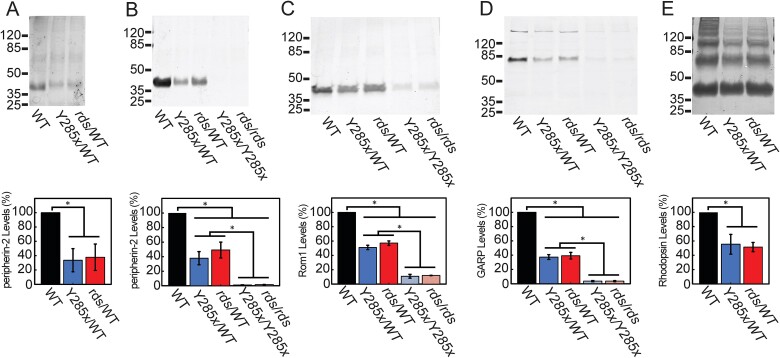
Mutations in PRPH2 are a relatively common cause of sight-robbing inherited retinal degenerations (IRDs). Peripherin-2 (PRPH2) is a photoreceptor-specific tetraspanin protein that structures the disk rim membranes of rod and cone outer segment (OS) organelles, and is required for OS morphogenesis. PRPH2 is noteworthy for its broad spectrum of disease phenotypes; both inter- and intra-familial heterogeneity have been widely observed and this variability in disease expression and penetrance confounds efforts to understand genotype-phenotype correlations and pathophysiology. Here we report the generation and initial characterization of a gene-edited animal model for PRPH2 disease associated with a nonsense mutation (c.1095:C>A, p.Y285X), which is predicted to truncate the peripherin-2 C-terminal domain. Young (P21) Prph2Y285X/WT mice developed near-normal photoreceptor numbers; however, OS membrane architecture was disrupted, OS protein levels were reduced, and in vivo and ex vivo electroretinography (ERG) analyses found that rod and cone photoreceptor function were each severely reduced. Interestingly, ERG studies also revealed that rod-mediated downstream signaling (b-waves) were functionally compensated in the young animals. This resiliency in retinal function was retained at P90, by which time substantial IRD-related photoreceptor loss had occurred. Altogether, the current studies validate a new mouse model for investigating PRPH2 disease pathophysiology, and demonstrate that rod and cone photoreceptor function and structure are each directly and substantially impaired by the Y285X mutation. They also reveal that Prph2 mutations can induce a functional compensation that resembles homeostatic plasticity, which can stabilize rod-derived signaling, and potentially dampen retinal dysfunction during some PRPH2-associated IRDs.
PRPH2 基因突变是导致致盲遗传性视网膜变性(IRDs)的一个相对常见的原因。外周蛋白-2(PRPH2)是一种光感受器特异性四跨膜蛋白,它构建了视杆和视锥外节(OS)细胞器的盘缘膜,是 OS 形态发生所必需的。PRPH2 的疾病表型谱很广,这一点值得注意;已经广泛观察到了种内和种间的异质性,这种疾病表型的变异性使理解基因型-表型相关性和病理生理学的努力变得复杂。在这里,我们报告了一种与无义突变(c.1095:C>A,p.Y285X)相关的 PRPH2 疾病的基因编辑动物模型的产生和初步特征,该突变预计会截断外周蛋白-2 的 C 末端结构域。年轻(P21)Prph2Y285X/WT 小鼠的视锥细胞数量发育正常;然而,OS 膜结构被破坏,OS 蛋白水平降低,体内和体外视网膜电图(ERG)分析发现视杆和视锥光感受器功能均严重降低。有趣的是,ERG 研究还表明,年轻动物的视杆介导的下游信号(b-波)在功能上得到了补偿。这种视网膜功能的弹性在 P90 时得以保留,此时已经发生了大量与 IRD 相关的光感受器损失。总的来说,目前的研究为研究 PRPH2 疾病的病理生理学提供了一个新的小鼠模型,并表明 Y285X 突变直接且严重地损害了视杆和视锥光感受器的功能和结构。它们还表明,Prph2 突变可以诱导一种类似于稳态可塑性的功能补偿,这种补偿可以稳定视杆源性信号,并在某些与 PRPH2 相关的 IRD 中潜在地减轻视网膜功能障碍。