LAMSADE, Université Paris-Dauphine, Pl. du Maréchal de Lattre de Tassigny, 75016 Paris, France.
Randall Centre for Cell and Molecular Biophysics, School of Basic and Medical Biosciences, King's College London, London SE1 1UL, United Kingdom.
J Chem Inf Model. 2024 Sep 23;64(18):7097-7107. doi: 10.1021/acs.jcim.4c01451. Epub 2024 Sep 9.
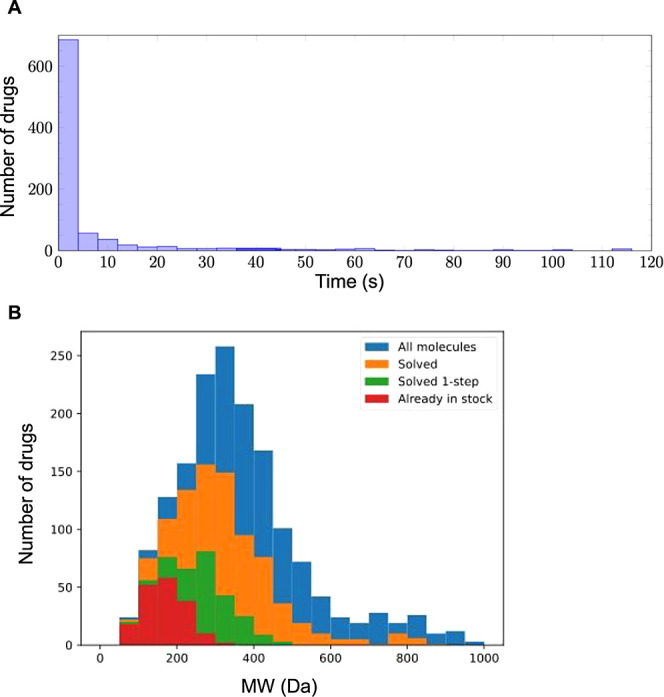
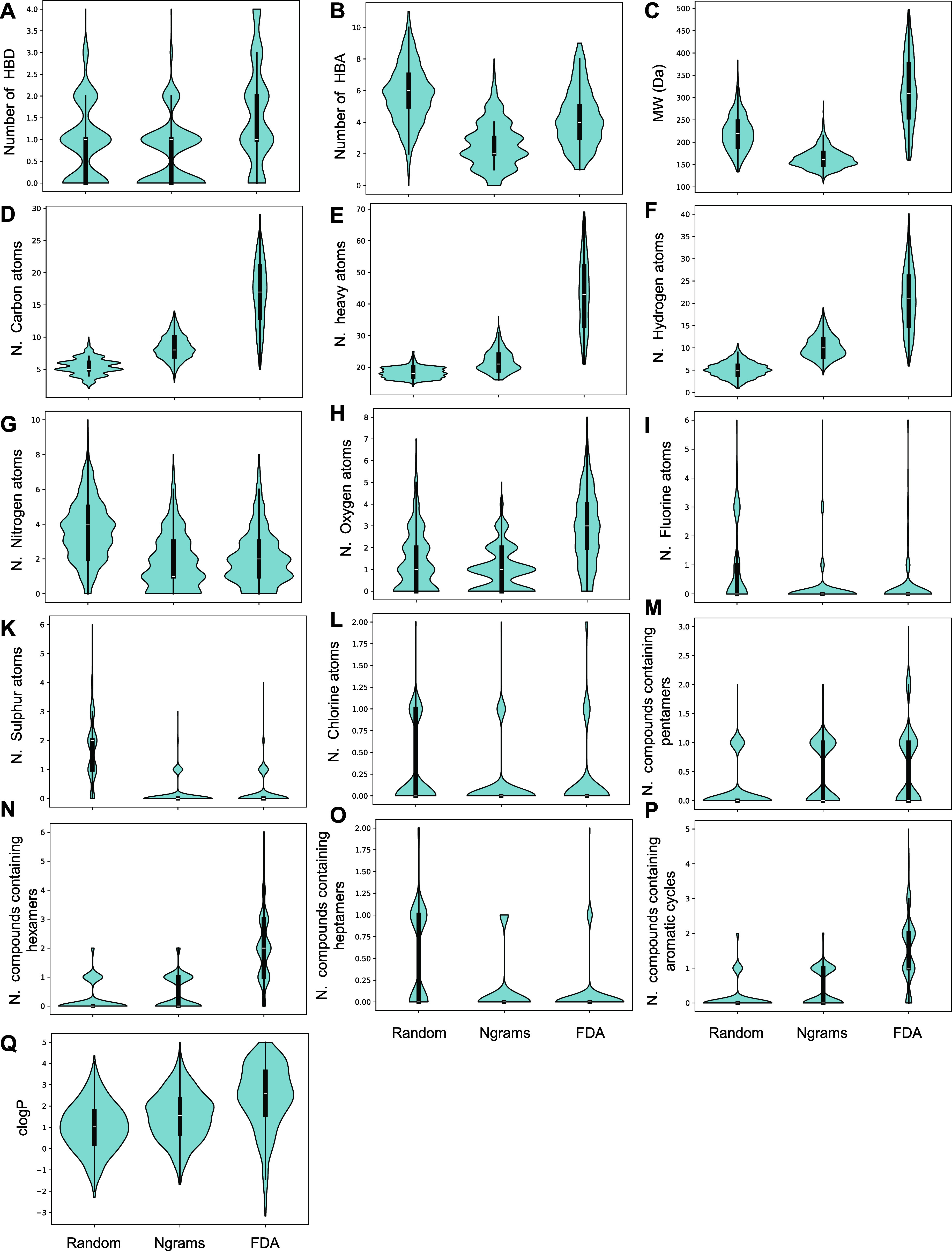
A growing number of deep learning (DL) methodologies have recently been developed to design novel compounds and expand the chemical space within virtual libraries. Most of these neural network approaches design molecules to specifically bind a target based on its structural information and/or knowledge of previously identified binders. Fewer attempts have been made to develop approaches for design of virtual libraries, as synthesizability of generated molecules remains a challenge. In this work, we developed a new Monte Carlo Search (MCS) algorithm, DrugSynthMC (Dru Synthesis using Monte Carlo), in conjunction with DL and statistical-based priors to generate thousands of interpretable chemical structures and novel drug-like molecules per second. DrugSynthMC produces drug-like compounds using an atom-based search model that builds molecules as SMILES, character by character. Designed molecules follow Lipinski's "rule of 5″, show a high proportion of highly water-soluble nontoxic predicted-to-be synthesizable compounds, and efficiently expand the chemical space within the libraries, without reliance on training data sets, synthesizability metrics, or enforcing during SMILES generation. Our approach can function with or without an underlying neural network and is thus easily explainable and versatile. This ease in drug-like molecule generation allows for future integration of score functions aimed at different target- or job-oriented goals. Thus, DrugSynthMC is expected to enable the functional assessment of large compound libraries covering an extensive novel chemical space, overcoming the limitations of existing drug collections. The software is available at https://github.com/RoucairolMilo/DrugSynthMC.
最近,越来越多的深度学习 (DL) 方法被开发出来,用于设计新型化合物并扩展虚拟库中的化学空间。这些神经网络方法中的大多数都是根据目标的结构信息和/或先前鉴定的配体的知识来设计专门与目标结合的分子。而设计虚拟库的方法则较少尝试,因为生成分子的可合成性仍然是一个挑战。在这项工作中,我们开发了一种新的蒙特卡罗搜索 (MCS) 算法 DrugSynthMC(使用蒙特卡罗的药物合成),结合了 DL 和基于统计的先验知识,每秒可以生成数千个可解释的化学结构和新型类药性分子。DrugSynthMC 使用基于原子的搜索模型来生成类药性化合物,该模型逐字符构建分子的 SMILES 表示。设计的分子符合 Lipinski 的“五规则”,表现出高比例的高水溶性、无毒、预测可合成的化合物,并且有效地扩展了库内的化学空间,无需依赖训练数据集、可合成性度量或在 SMILES 生成过程中强制执行。我们的方法可以在没有或有基础神经网络的情况下运行,因此易于解释和多功能。这种生成类药性分子的简便性允许未来集成针对不同目标或面向任务的目标的评分函数。因此,DrugSynthMC 有望实现对涵盖广泛新化学空间的大型化合物库的功能评估,克服现有药物库的局限性。该软件可在 https://github.com/RoucairolMilo/DrugSynthMC 上获得。