Wenz Brandon M, He Yuan, Chen Nae-Chyun, Pickrell Joseph K, Li Jeremiah H, Dudek Max F, Li Taibo, Keener Rebecca, Voight Benjamin F, Brown Christopher D, Battle Alexis
Genetics and Epigenetics Program, Cell and Molecular Biology Graduate Group, Biomedical Graduate Studies, University of Pennsylvania - Perelman School of Medicine, Philadelphia PA 19104.
Department of Biomedical Engineering, Johns Hopkins University; Baltimore, MD, 21218.
bioRxiv. 2024 Sep 5:2024.09.04.610850. doi: 10.1101/2024.09.04.610850.
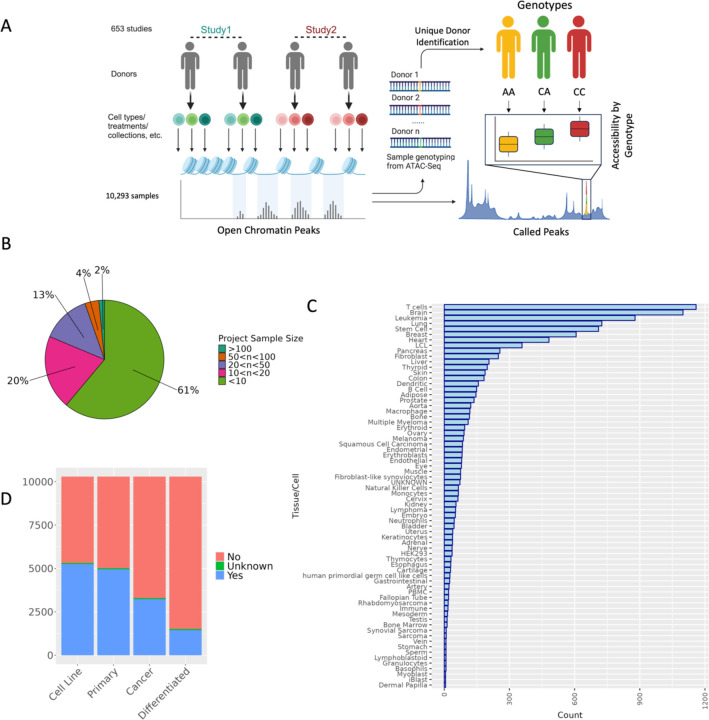
Understanding the genetic causes for variability in chromatin accessibility can shed light on the molecular mechanisms through which genetic variants may affect complex traits. Thousands of ATAC-seq samples have been collected that hold information about chromatin accessibility across diverse cell types and contexts, but most of these are not paired with genetic information and come from diverse distinct projects and laboratories.
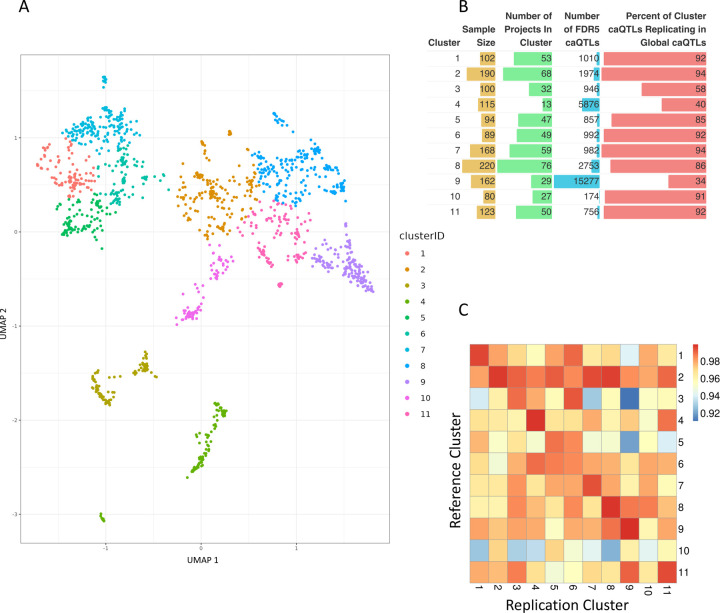
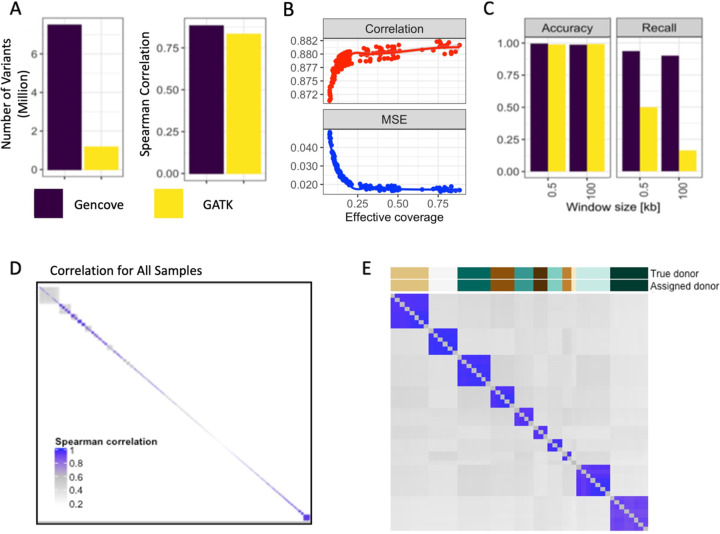
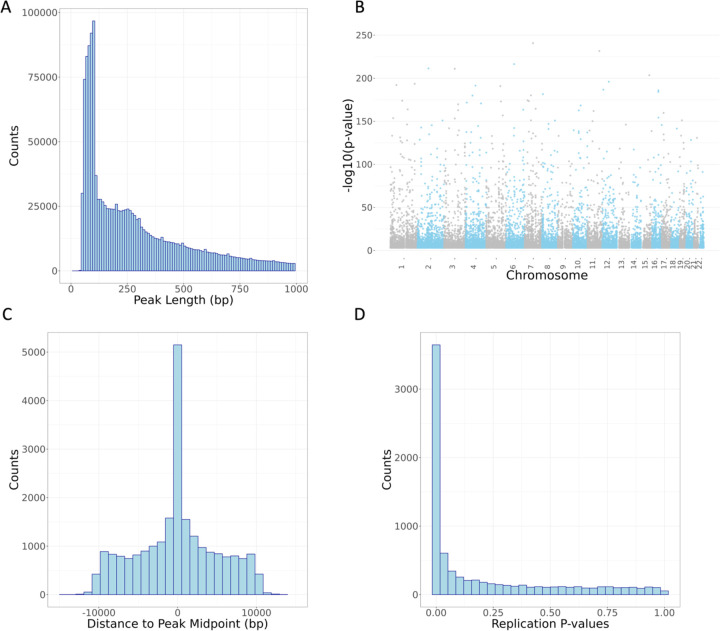
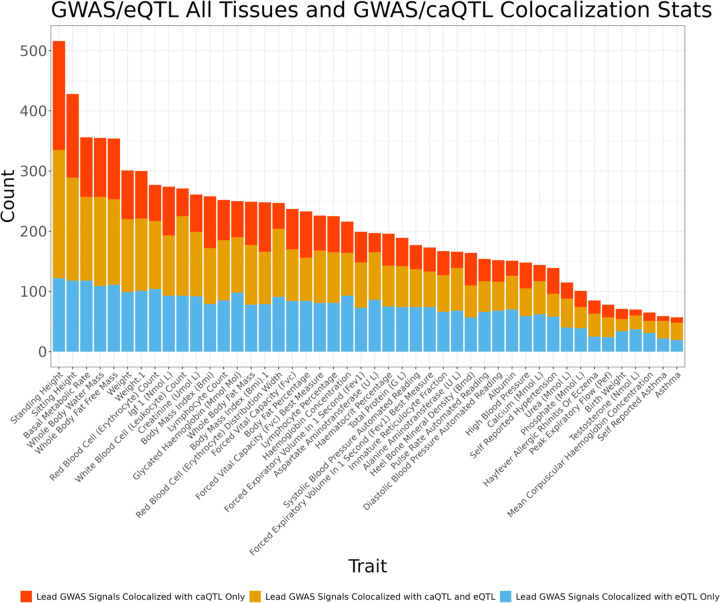
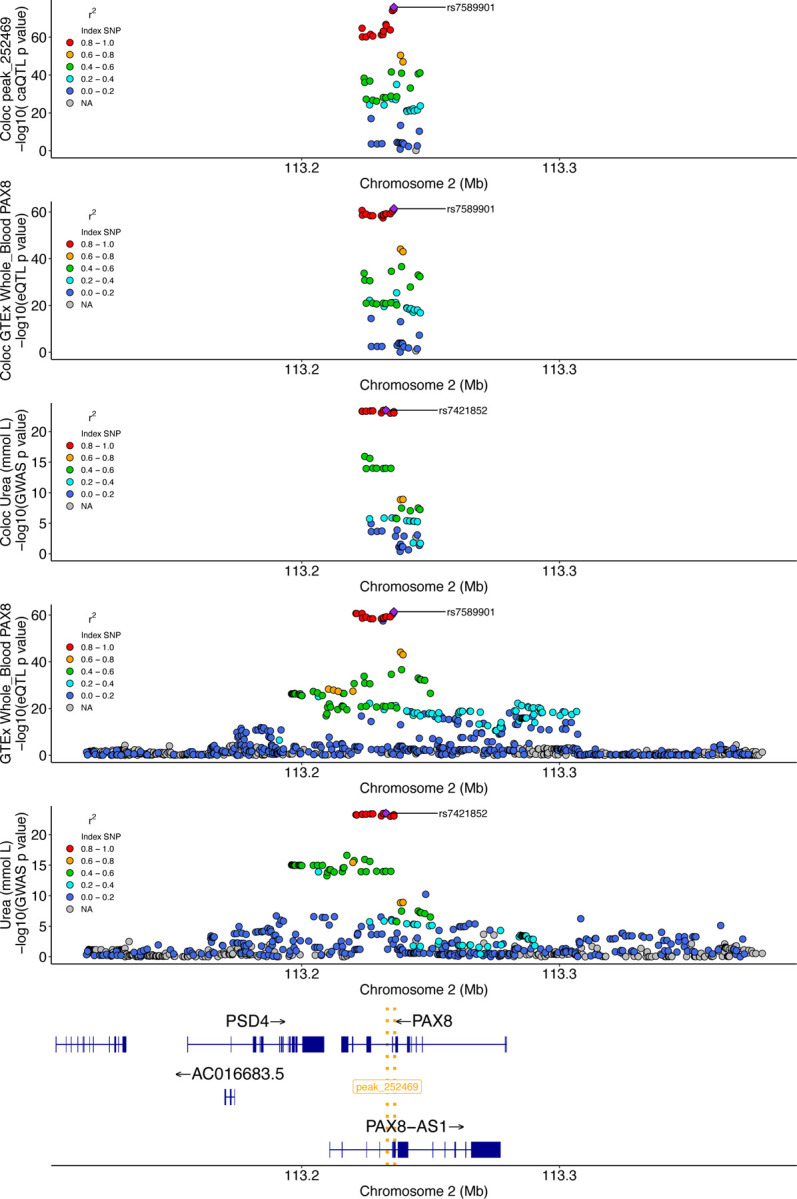
We report here joint genotyping, chromatin accessibility peak calling, and discovery of quantitative trait loci which influence chromatin accessibility (caQTLs), demonstrating the capability of performing caQTL analysis on a large scale in a diverse sample set without pre-existing genotype information. Using 10,293 profiling samples representing 1,454 unique donor individuals across 653 studies from public databases, we catalog 23,381 caQTLs in total. After joint discovery analysis, we cluster samples based on accessible chromatin profiles to identify context-specific caQTLs. We find that caQTLs are strongly enriched for annotations of gene regulatory elements across diverse cell types and tissues and are often strongly linked with genetic variation associated with changes in expression (eQTLs), indicating that caQTLs can mediate genetic effects on gene expression. We demonstrate sharing of causal variants for chromatin accessibility and diverse complex human traits, enabling a more complete picture of the genetic mechanisms underlying complex human phenotypes.
Our work provides a proof of principle for caQTL calling from previously ungenotyped samples, and represents one of the largest, most diverse caQTL resources currently available, informing mechanisms of genetic regulation of gene expression and contribution to disease.
了解染色质可及性变异的遗传原因,有助于揭示遗传变异可能影响复杂性状的分子机制。目前已收集了数千个ATAC-seq样本,这些样本包含了不同细胞类型和背景下染色质可及性的信息,但其中大多数未与遗传信息配对,且来自不同的项目和实验室。
我们在此报告了联合基因分型、染色质可及性峰识别以及影响染色质可及性的数量性状位点(caQTLs)的发现,证明了在没有预先存在的基因型信息的情况下,能够在多样化的样本集中大规模进行caQTL分析。利用来自公共数据库的653项研究中的10293个分析样本,这些样本代表了1454个独特的供体个体,我们总共编目了23381个caQTLs。经过联合发现分析后,我们根据可及染色质图谱对样本进行聚类,以识别特定背景下的caQTLs。我们发现,caQTLs在不同细胞类型和组织的基因调控元件注释中高度富集,并且通常与表达变化相关的遗传变异(eQTLs)紧密相连,这表明caQTLs可以介导对基因表达的遗传效应。我们证明了染色质可及性和多种复杂人类性状的因果变异存在共享,从而能够更全面地了解复杂人类表型背后的遗传机制。
我们的工作为从先前未进行基因分型的样本中识别caQTL提供了原理证明,并且代表了目前可用的最大、最多样化的caQTL资源之一,为基因表达的遗传调控机制及其对疾病的影响提供了信息。