Beijing Key Laboratory of the Innovative Development of Functional Staple and the Nutritional Intervention for Chronic Disease, Department of Clinical Nutrition, Peking Union Medical College Hospital (PUMCH), Beijing, 100730, China.
Department of Pediatrics, Peking Union Medical College Hospital (PUMCH), Beijing, 100730, China.
BMC Pediatr. 2024 Sep 28;24(1):603. doi: 10.1186/s12887-024-05054-w.
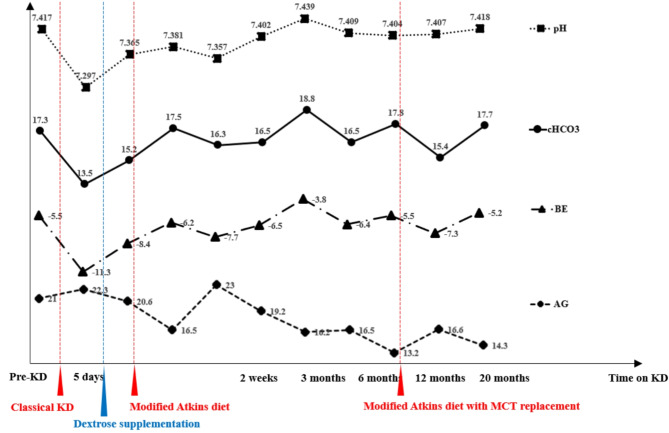
As a rare mitochondrial disorder, the pyruvate dehydrogenase complex (PDC) deficiency is a rare inborn disease characterized with glucose metabolism defects, which leads to neurological dysfunction, serum lactic acid buildup and a resultant trend of metabolic acidosis. Although the ketogenic diet (KD) is the first-line treatment for PDC deficiency, there is currently no widely accepted consensus on specific implementation of KD for this condition. Due to the combined effect of pre-existing hyperlactacidemia and KD-induced ketoacidosis that can further exacerbate metabolic disturbances, maintaining metabolic homeostasis should be prioritized during the implementation of KD.
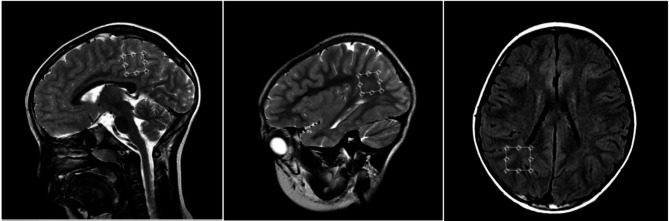
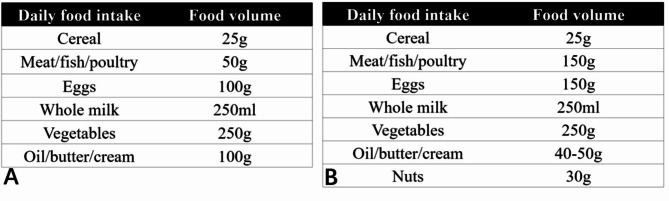
Herein, the authors present a 6-year-old boy with lactic acidosis, ataxia, hypotonia and neuromotor development retardation. The KD was started after the patient was diagnosed with PDC deficiency based on genetic testing. The initiation with classic KD resulted in severe non-diabetic ketoacidosis with elevated anion gap, which was promptly alleviated by dextrose supplementation and dietary modification to a less-restrictive KD. Long-term supervision demonstrated the efficacy of a modified KD in improving both clinical course and metabolic acidosis of the patient.
This rare case adds to the limited evidence of KD application in PDC deficiency, and provides valuable insights into the importance of reasonably lowering the ketogenic ratio of KD at the start of treatment to reduce the risk of metabolic acidosis.
丙酮酸脱氢酶复合物(PDC)缺陷是一种罕见的线粒体疾病,属于罕见的先天性疾病,其特征为葡萄糖代谢缺陷,导致神经功能障碍、血清乳酸堆积以及代谢性酸中毒的趋势。虽然生酮饮食(KD)是 PDC 缺陷的一线治疗方法,但目前对于这种疾病的 KD 具体实施尚无广泛接受的共识。由于预先存在的高乳酸血症和 KD 诱导的酮症酸中毒的联合作用可能进一步加重代谢紊乱,因此在实施 KD 时应优先维持代谢平衡。
本文报道了一例 6 岁男孩,表现为乳酸酸中毒、共济失调、肌张力低下和神经运动发育迟缓。在基因检测诊断为 PDC 缺陷后,开始使用 KD。经典 KD 的起始导致严重的非糖尿病酮症酸中毒,阴离子间隙升高,通过补充葡萄糖和饮食调整为限制较少的 KD 后迅速缓解。长期监测表明,改良 KD 可改善患者的临床病程和代谢性酸中毒。
本罕见病例增加了 KD 在 PDC 缺陷中的应用的有限证据,并提供了有价值的见解,即治疗开始时合理降低 KD 的生酮比以降低代谢性酸中毒的风险非常重要。