Department of Pulmonary and Critical Care Medicine, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, Shandong, 250021, China.
Shandong Provincial Key Medical and Health Laboratory of Cell Metabolism, Central Hospital Affiliated to Shandong First Medical University, Jinan, Shandong, 250021, China.
J Exp Clin Cancer Res. 2024 Sep 30;43(1):268. doi: 10.1186/s13046-024-03181-x.
Metabolic reprogramming plays a pivotal role in tumorigenesis and development of lung adenocarcinoma (LUAD). However, the precise mechanisms and potential targets for metabolic reprogramming in LUAD remain elusive. Our prior investigations revealed that the mitochondrial ribosomal protein MRPL12, identified as a novel mitochondrial transcriptional regulatory gene, exerts a critical influence on mitochondrial metabolism. Despite this, the role and regulatory mechanisms underlying MRPL12's transcriptional activity in cancers remain unexplored.
Human LUAD tissues, Tp53;Kras-driven LUAD mouse models, LUAD patient-derived organoids (PDO), and LUAD cell lines were used to explored the expression and function of MRPL12. The posttranslational modification of MRPL12 was analyzed by mass spectrometry, and the oncogenic role of key phosphorylation sites of MRPL12 in LUAD development was verified in vivo and in vitro.
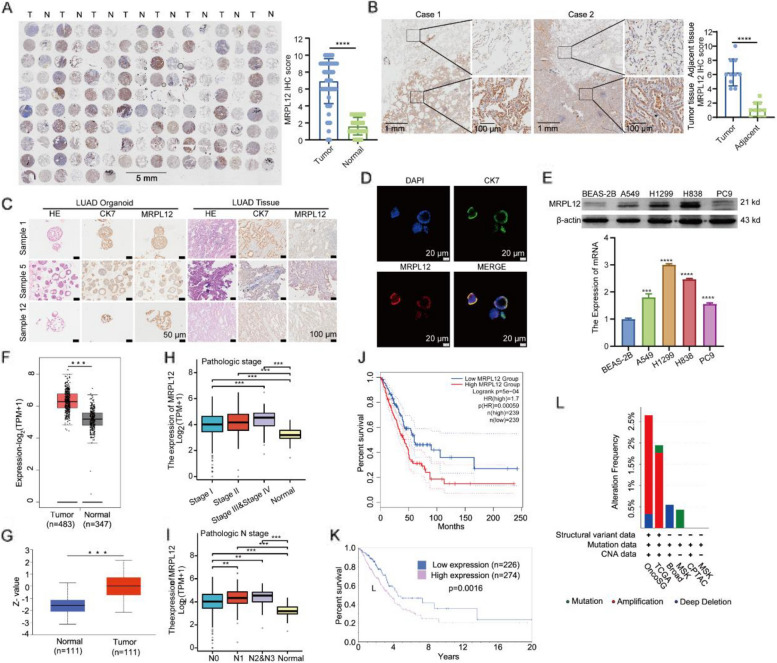
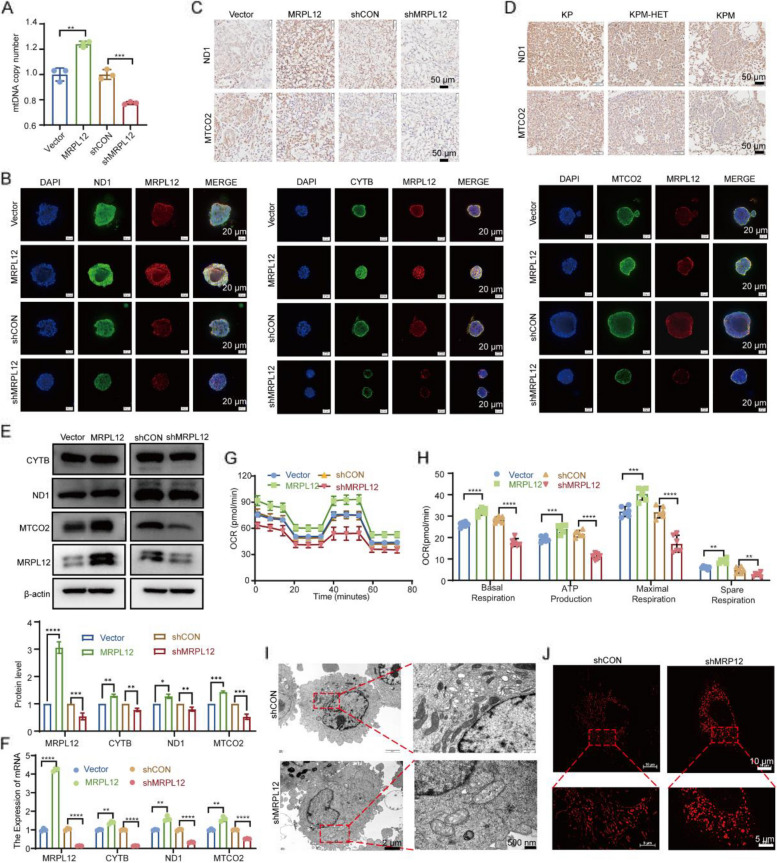
MRPL12 was upregulated in human LUAD tissues, Tp53;Kras-driven LUAD tissues in mice, LUAD PDO, and LUAD cell lines, correlating with poor patient survival. Overexpression of MRPL12 significantly promoted LUAD tumorigenesis, metastasis, and PDO formation, while MRPL12 knockdown elicited the opposite phenotype. Additionally, MRPL12 deletion in a Tp53;Kras-driven mouse LUAD model conferred a notable survival advantage, delaying tumor onset and reducing malignant progression. Mechanistically, we discovered that MRPL12 promotes tumor progression by upregulating mitochondrial oxidative phosphorylation. Furthermore, we identified UBASH3B as a specific binder of MRPL12, dephosphorylating tyrosine 60 in MRPL12 (MRPL12 Y60) and inhibiting its oncogenic functions. The decrease in MRPL12 Y60 phosphorylation impeded the binding of MRPL12 to POLRMT, downregulating mitochondrial metabolism in LUAD cells. In-depth in vivo, in vitro, and organoid models validated the inhibitory effect of MRPL12 Y60 mutation on LUAD.
This study establishes MRPL12 as a novel oncogene in LUAD, contributing to LUAD pathogenesis by orchestrating mitochondrial metabolism reprogramming towards oxidative phosphorylation (OXPHOS). Furthermore, it confirms Y60 as a specific phosphorylation modification site regulating MRPL12's oncogenic functions, offering insights for the development of LUAD-specific targeted drugs and clinical interventions.
代谢重编程在肺腺癌(LUAD)的发生和发展中起着关键作用。然而,LUAD 中代谢重编程的确切机制和潜在靶点仍不清楚。我们之前的研究表明,作为一种新的线粒体转录调控基因的线粒体核糖体蛋白 MRPL12 对线粒体代谢有重要影响。尽管如此,MRPL12 的转录活性在癌症中的作用和调节机制仍未被探索。
使用人 LUAD 组织、Tp53;Kras 驱动的 LUAD 小鼠模型、LUAD 患者来源的类器官(PDO)和 LUAD 细胞系来研究 MRPL12 的表达和功能。通过质谱分析了 MRPL12 的翻译后修饰,并在体内和体外验证了 MRPL12 关键磷酸化位点在 LUAD 发展中的致癌作用。
MRPL12 在人 LUAD 组织、Tp53;Kras 驱动的 LUAD 小鼠组织、LUAD PDO 和 LUAD 细胞系中上调,与患者预后不良相关。MRPL12 的过表达显著促进了 LUAD 肿瘤的发生、转移和 PDO 的形成,而 MRPL12 的敲低则产生了相反的表型。此外,Tp53;Kras 驱动的 LUAD 小鼠模型中 MRPL12 的缺失赋予了显著的生存优势,延迟了肿瘤的发生并减少了恶性进展。机制上,我们发现 MRPL12 通过上调线粒体氧化磷酸化来促进肿瘤的进展。此外,我们发现 UBASH3B 是 MRPL12 的特异性结合物,使 MRPL12 的酪氨酸 60 去磷酸化(MRPL12 Y60)并抑制其致癌功能。MRPL12 Y60 磷酸化的减少阻碍了 MRPL12 与 POLRMT 的结合,下调了 LUAD 细胞中的线粒体代谢。在深入的体内、体外和类器官模型中验证了 MRPL12 Y60 突变对 LUAD 的抑制作用。
本研究确立了 MRPL12 是 LUAD 中的一种新的癌基因,通过协调向氧化磷酸化(OXPHOS)的线粒体代谢重编程促进 LUAD 的发病机制。此外,它证实了 Y60 是调节 MRPL12 致癌功能的特定磷酸化修饰位点,为 LUAD 特异性靶向药物的开发和临床干预提供了新的思路。