Lundgren Kristoffer J M, Caldararu Octav, Oksanen Esko, Ryde Ulf
Department of Computational Chemistry, Lund University, Chemical Centre, PO Box 124, SE-221 00 Lund, Sweden.
IUCrJ. 2024 Nov 1;11(Pt 6):921-937. doi: 10.1107/S2052252524008406.
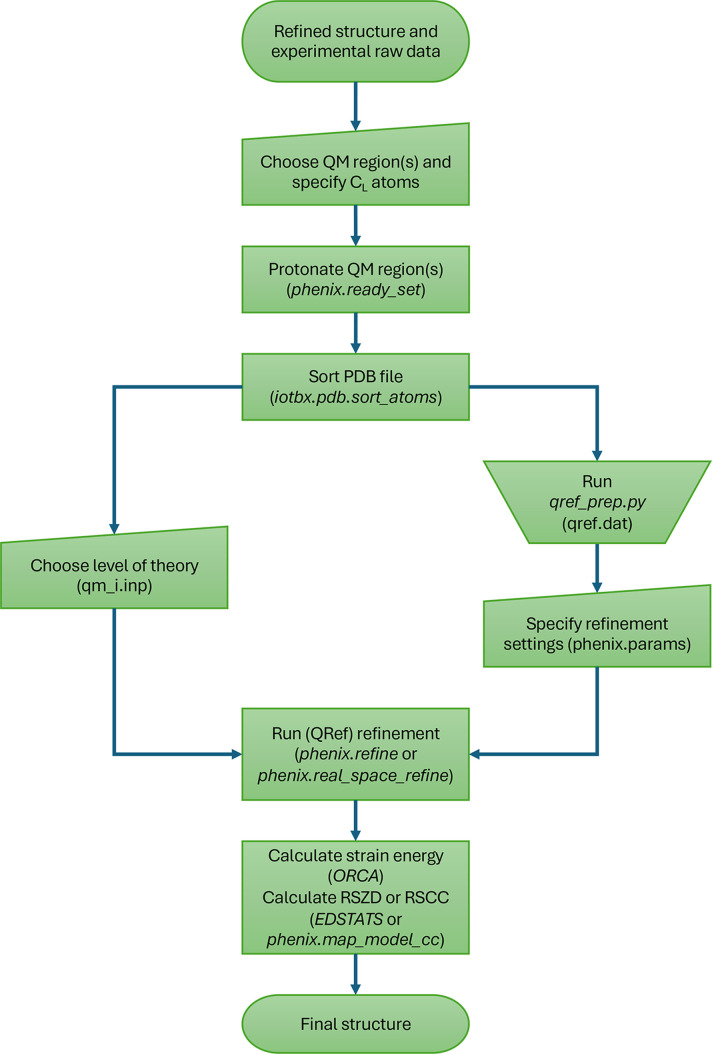
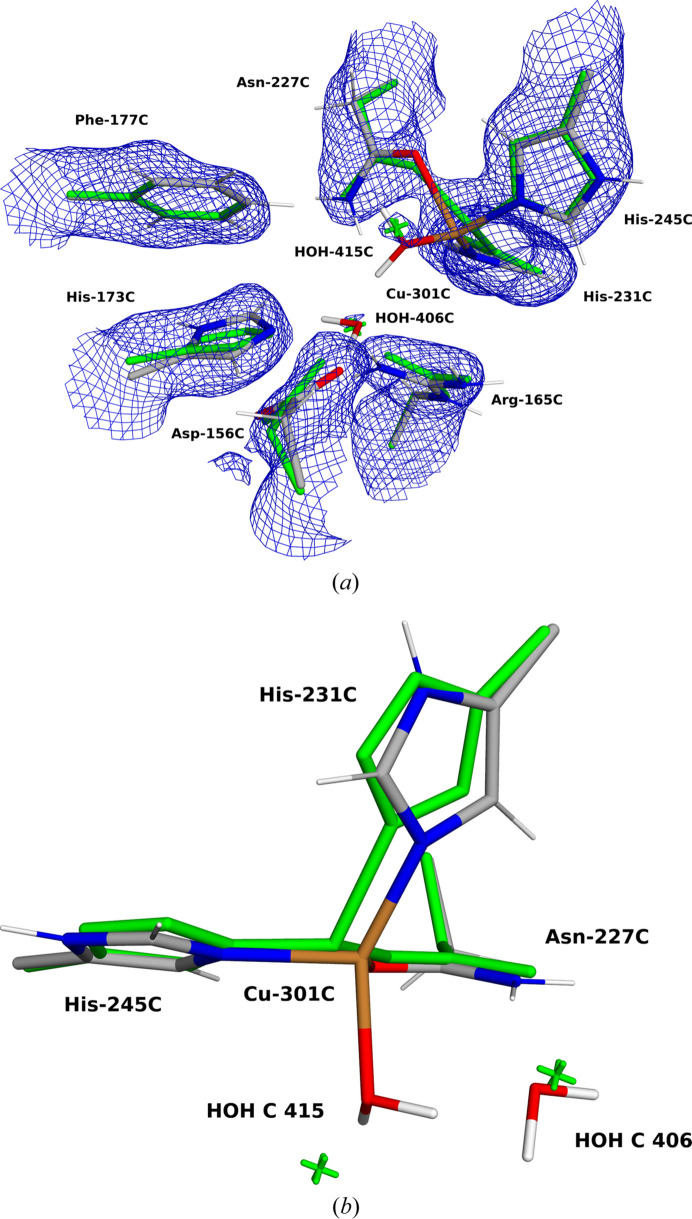
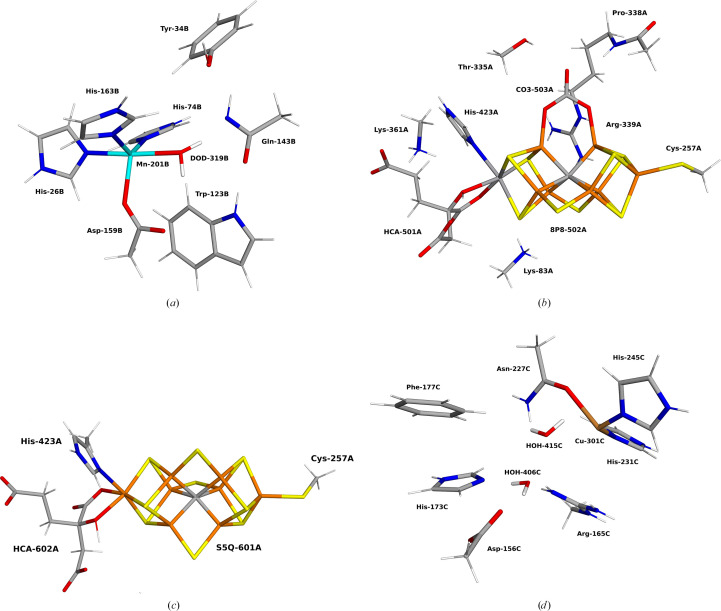
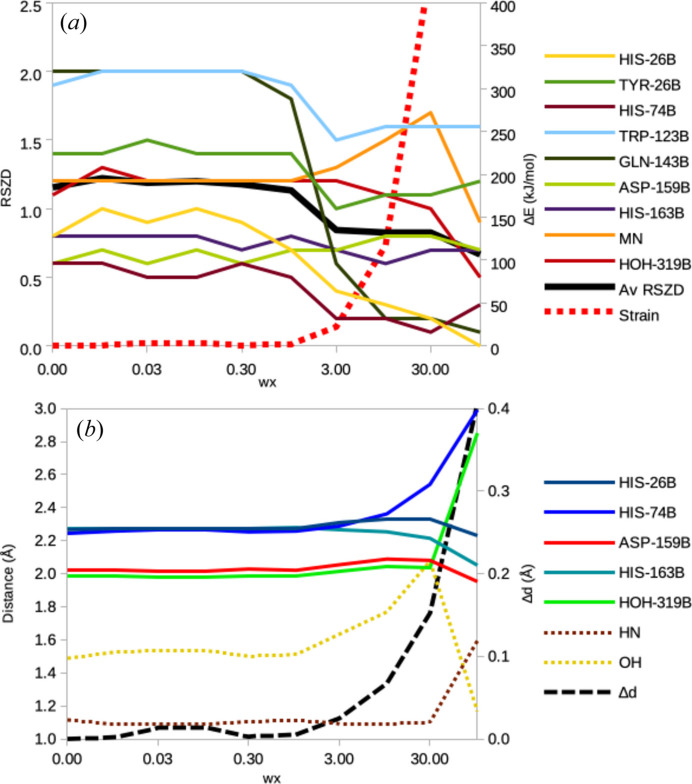
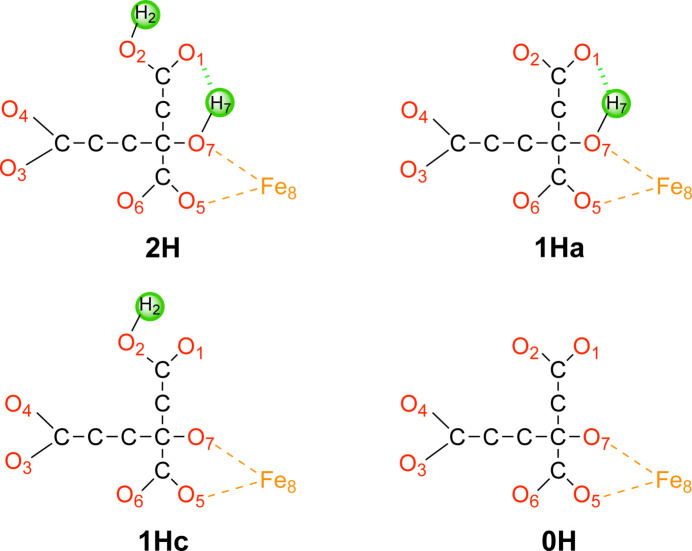
X-ray and neutron crystallography, as well as cryogenic electron microscopy (cryo-EM), are the most common methods to obtain atomic structures of biological macromolecules. A feature they all have in common is that, at typical resolutions, the experimental data need to be supplemented by empirical restraints, ensuring that the final structure is chemically reasonable. The restraints are accurate for amino acids and nucleic acids, but often less accurate for substrates, inhibitors, small-molecule ligands and metal sites, for which experimental data are scarce or empirical potentials are harder to formulate. This can be solved using quantum mechanical calculations for a small but interesting part of the structure. Such an approach, called quantum refinement, has been shown to improve structures locally, allow the determination of the protonation and oxidation states of ligands and metals, and discriminate between different interpretations of the structure. Here, we present a new implementation of quantum refinement interfacing the widely used structure-refinement software Phenix and the freely available quantum mechanical software ORCA. Through application to manganese superoxide dismutase and V- and Fe-nitrogenase, we show that the approach works effectively for X-ray and neutron crystal structures, that old results can be reproduced and structural discrimination can be performed. We discuss how the weight factor between the experimental data and the empirical restraints should be selected and how quantum mechanical quality measures such as strain energies should be calculated. We also present an application of quantum refinement to cryo-EM data for particulate methane monooxygenase and show that this may be the method of choice for metal sites in such structures because no accurate empirical restraints are currently available for metals.
X射线晶体学、中子晶体学以及低温电子显微镜技术(cryo-EM)是获取生物大分子原子结构最常用的方法。它们共有的一个特点是,在典型分辨率下,实验数据需要用经验性约束进行补充,以确保最终结构在化学上合理。这些约束对氨基酸和核酸是准确的,但对于底物、抑制剂、小分子配体和金属位点往往不太准确,因为针对这些的实验数据稀缺或经验势更难制定。这可以通过对结构中一个小但有趣的部分进行量子力学计算来解决。这种方法称为量子精修,已被证明可以局部改善结构,确定配体和金属的质子化和氧化态,并区分结构的不同解释。在这里,我们展示了量子精修的一种新实现方式,它将广泛使用的结构精修软件Phenix与免费的量子力学软件ORCA连接起来。通过应用于锰超氧化物歧化酶以及钒和铁固氮酶,我们表明该方法对X射线和中子晶体结构有效,可以重现旧结果并进行结构区分。我们讨论了应如何选择实验数据和经验性约束之间的权重因子,以及应如何计算诸如应变能等量子力学质量度量。我们还展示了量子精修在颗粒状甲烷单加氧酶低温电子显微镜数据中的应用,并表明这可能是此类结构中金属位点的首选方法,因为目前对于金属尚无准确的经验性约束。