Science Institute, University of Iceland, Dunhagi 3, 107 Reykjavik, Iceland.
Max-Planck Institute for Chemical Energy Conversion, Stiftstrasse 34-36, 45470 Mülheim an der Ruhr, Germany.
J Chem Theory Comput. 2022 Mar 8;18(3):1437-1457. doi: 10.1021/acs.jctc.1c00753. Epub 2022 Feb 15.
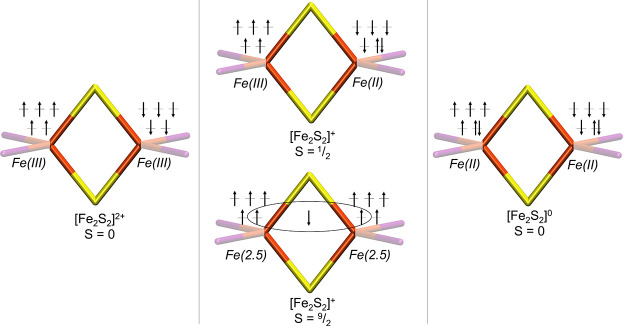
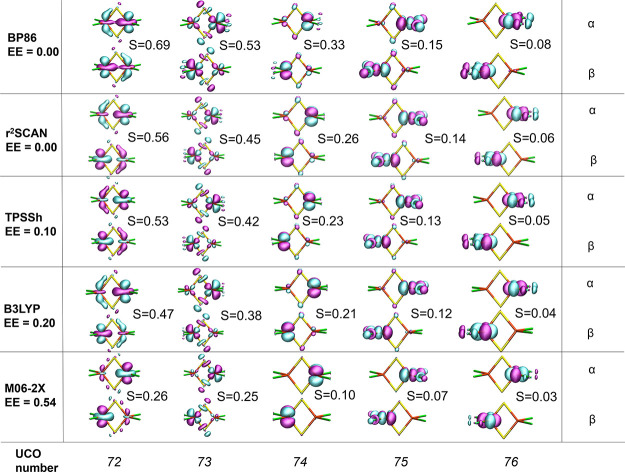
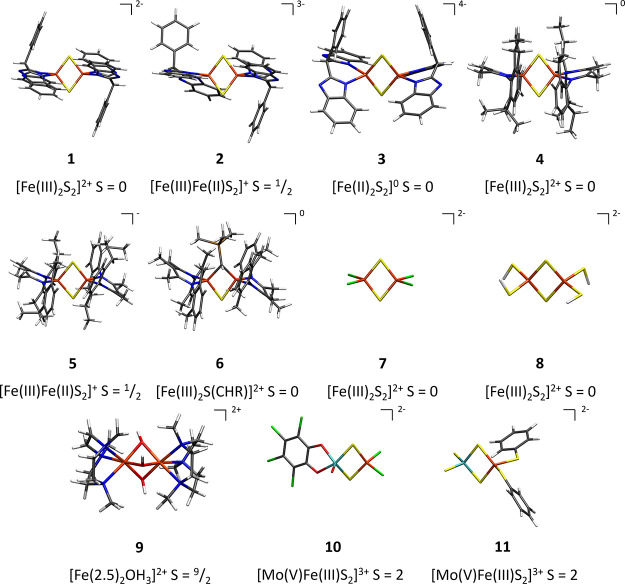
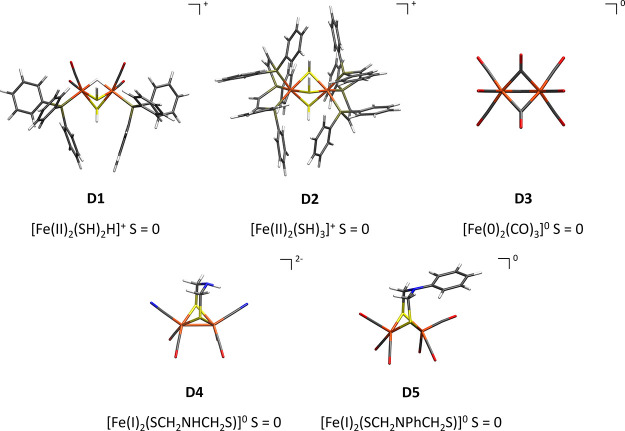
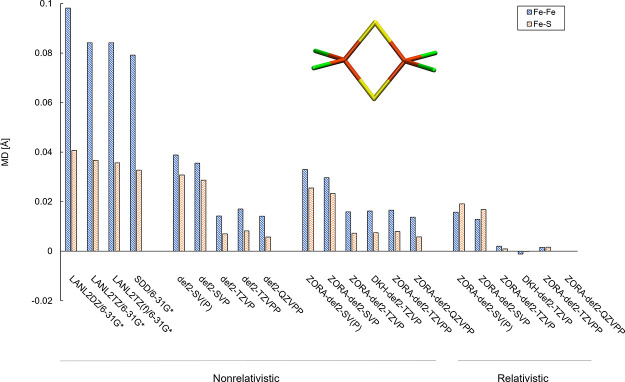
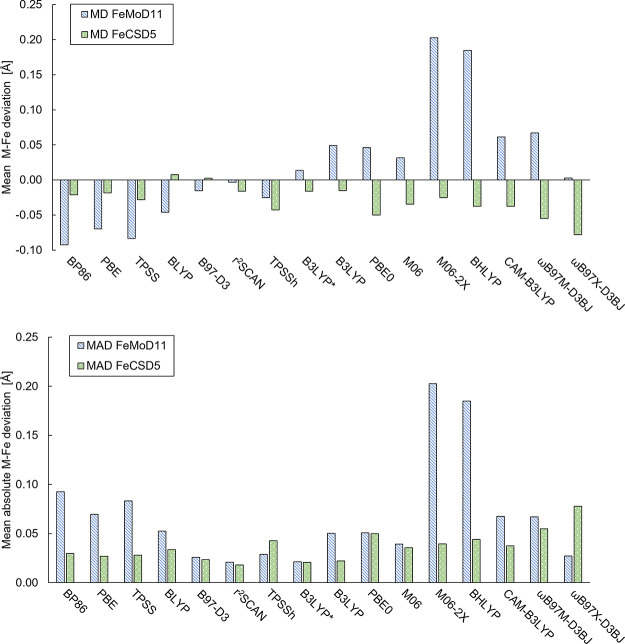
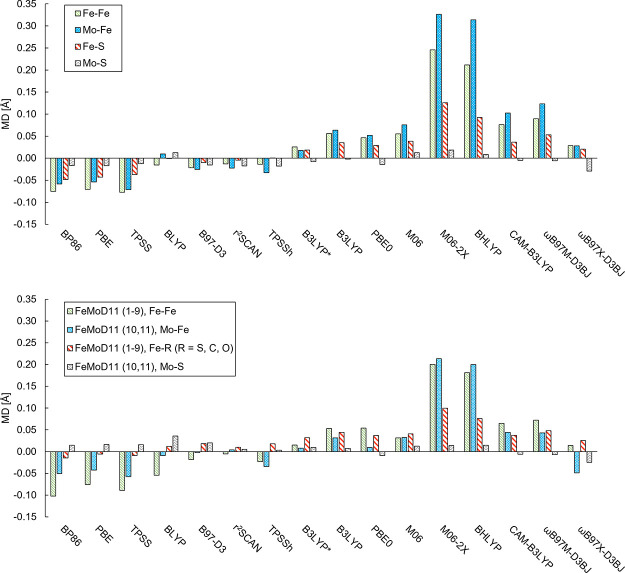
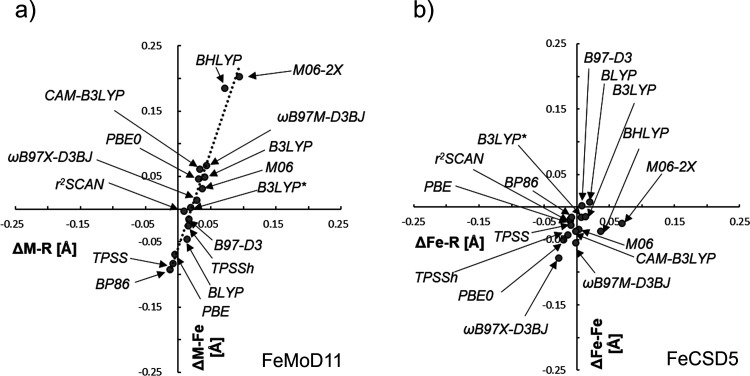
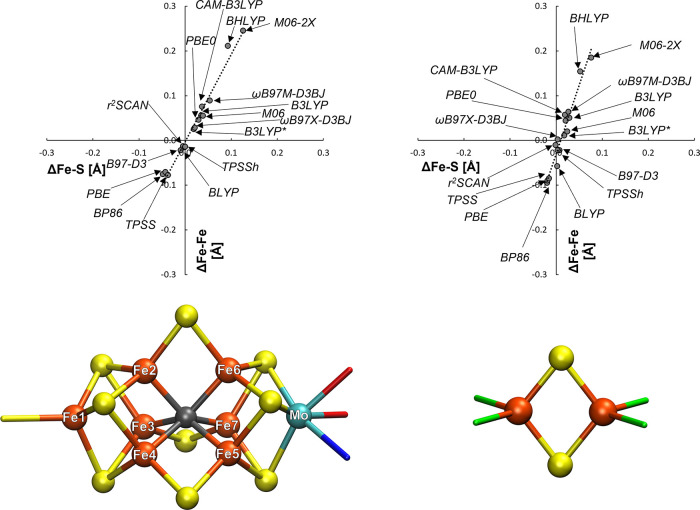
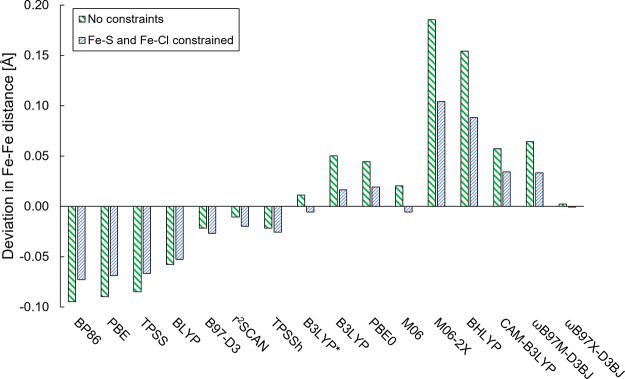
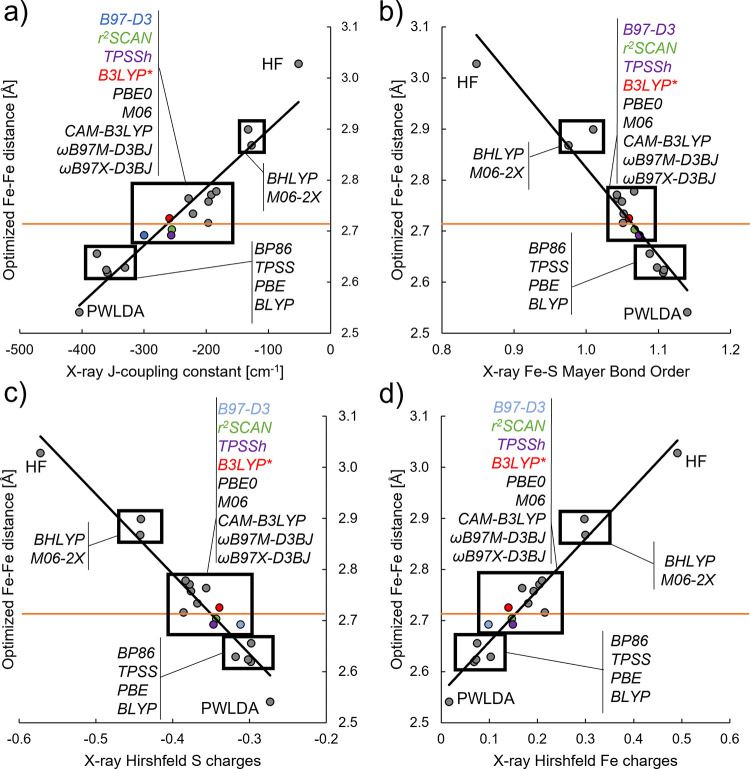
The open-shell electronic structure of iron-sulfur clusters presents considerable challenges to quantum chemistry, with the complex iron-molybdenum cofactor (FeMoco) of nitrogenase representing perhaps the ultimate challenge for either wavefunction or density functional theory. While broken-symmetry density functional theory has seen some success in describing the electronic structure of such cofactors, there is a large exchange-correlation functional dependence in calculations that is not fully understood. In this work, we present a geometric benchmarking test set, FeMoD11, of synthetic spin-coupled Fe-Fe and Mo-Fe dimers, with relevance to the molecular and electronic structure of the Mo-nitrogenase FeMo cofactor. The reference data consists of high-resolution crystal structures of metal dimer compounds in different oxidation states. Multiple density functionals are tested on their ability to reproduce the local geometry, specifically the Fe-Fe/Mo-Fe distance, for both antiferromagnetically coupled and ferromagnetically coupled dimers via the broken-symmetry approach. The metal-metal distance is revealed not only to be highly sensitive to the amount of exact exchange in the functional but also to the specific exchange and correlation functionals. For the antiferromagnetically coupled dimers, the calculated metal-metal distance correlates well with the covalency of the bridging metal-ligand bonds, as revealed via the corresponding orbital analysis, Hirshfeld S/Fe charges, and Fe-S Mayer bond order. Superexchange via bridging ligands is expected to be the dominant interaction in these dimers, and our results suggest that functionals that predict accurate Fe-Fe and Mo-Fe distances describe the overall metal-ligand covalency more accurately and in turn the superexchange of these systems. The best performing density functionals of the 16 tested for the FeMoD11 test set are revealed to be either the nonhybrid functionals rSCAN and B97-D3 or hybrid functionals with 10-15% exact exchange: TPSSh and B3LYP*. These same four functionals are furthermore found to reproduce the high-resolution X-ray structure of FeMoco well according to quantum mechanics/molecular mechanics (QM/MM) calculations. Almost all nonhybrid functionals systematically underestimate Fe-Fe and Mo-Fe distances (with rSCAN and B97-D3 being the sole exceptions), while hybrid functionals with >15% exact exchange (including range-separated hybrid functionals) overestimate them. The results overall suggest rSCAN, B97-D3, TPSSh, and B3LYP* as accurate density functionals for describing the electronic structure of iron-sulfur clusters in general, including the complex FeMoco cluster of nitrogenase.
铁硫簇的开壳电子结构对量子化学提出了相当大的挑战,而氮酶的复杂铁钼辅因子(FeMoco)可能是对波函数或密度泛函理论的终极挑战。尽管破对称密度泛函理论在描述此类辅因子的电子结构方面取得了一些成功,但在计算中存在很大的交换相关泛函依赖性,这尚未完全理解。在这项工作中,我们提出了一个合成自旋偶联的 Fe-Fe 和 Mo-Fe 二聚体的几何基准测试集 FeMoD11,这与 Mo-氮酶 FeMo 辅因子的分子和电子结构有关。参考数据由不同氧化态的金属二聚体化合物的高分辨率晶体结构组成。通过破对称方法,测试了多种密度泛函在反铁磁耦合和铁磁耦合二聚体中重现局部几何形状(特别是 Fe-Fe/Mo-Fe 距离)的能力。金属-金属距离不仅对功能中精确交换的数量高度敏感,而且对特定的交换和相关泛函也敏感。对于反铁磁耦合二聚体,计算出的金属-金属距离与桥接金属-配体键的共价性密切相关,这可以通过相应的轨道分析、Hirshfeld S/Fe 电荷和 Fe-S Mayer 键序揭示出来。预计通过桥接配体的超交换将是这些二聚体中的主要相互作用,我们的结果表明,预测准确的 Fe-Fe 和 Mo-Fe 距离的泛函更准确地描述了整个金属-配体的共价性,并相应地描述了这些系统的超交换。对于 FeMoD11 测试集,16 个测试的密度泛函中表现最好的是非杂化函数 rSCAN 和 B97-D3,或者是具有 10-15%精确交换的混合函数:TPSSh 和 B3LYP*。根据量子力学/分子力学(QM/MM)计算,这四种功能同样可以很好地再现 FeMoco 的高分辨率 X 射线结构。几乎所有非杂化函数都系统地低估了 Fe-Fe 和 Mo-Fe 距离(rSCAN 和 B97-D3 是唯一的例外),而具有>15%精确交换的混合函数(包括分段混合函数)则高估了它们。总体而言,结果表明 rSCAN、B97-D3、TPSSh 和 B3LYP* 是描述铁硫簇电子结构的准确密度泛函,包括氮酶的复杂 FeMoco 簇。