Paz Mercedes, Franco-Trecu Valentina, Szteren Diana, Costábile Alicia, Portela Cecilia, Bruno Alfredo, Moratorio Gonzalo, Moreno Pilar, Cristina Juan
Laboratorio de Virología Molecular, Centro de Investigaciones Nucleares, Facultad de Ciencias, Universidad de la República, Igua 4225, Montevideo 11400, Uruguay; Laboratorio de Evolución Experimental de Virus, Institut Pasteur de Montevideo, Mataojo 2020, Montevideo 11400, Uruguay; Centro de Innovación en Vigilancia Epidemiológica, Institut Pasteur Montevideo, Mataojo 2020, Montevideo 11400, Uruguay.
Departamento de Ecología y Evolución, Facultad de Ciencias, Igua 4224, 11400 Montevideo, Uruguay.
Virus Res. 2024 Dec;350:199472. doi: 10.1016/j.virusres.2024.199472. Epub 2024 Oct 5.
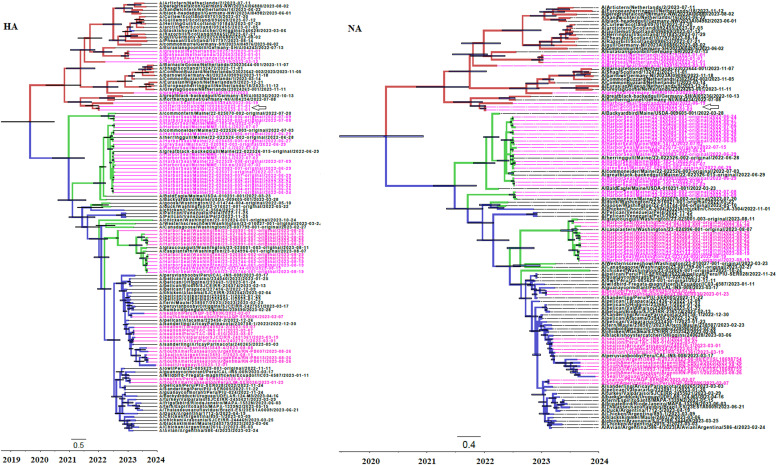
Highly pathogenic influenza A virus (HPIAV) H5N1 within the genetic clade 2.3.4.4b has emerged in wild birds in different regions of the world, leading to the death of >70 million birds. When these strains spread to pinniped species a remarkable mortality has also been observed. A detailed genetic characterization of HPIAV isolated from pinnipeds is essential to understand the potential spread of these viruses to other mammalian species, including humans. To gain insight into these matters a detailed phylogenetic analysis of HPIAV H5N1 2.3.4.4b strains isolated from pinniped species was performed. The results of these studies revealed multiple transmission events from birds to pinnipeds in all world regions. Different evolutionary histories of different genes of HPIAV H5N1 2.3.4.4b strains gave rise to the viruses infecting pinnipeds in different regions of the world. European strains isolated from pinnipeds represent a completely different genetic lineage from strains isolated from South American ones. All strains isolated from pinnipeds bear characteristics of a highly pathogenic form for of avian influenza in poultry. Amino acid substitutions, previously shown to confer an adaptive advantage for infecting mammals, were observed in different genes in all pinniped species studied.
高致病性甲型流感病毒(HPIAV)H5N1的2.3.4.4b遗传分支已在世界不同地区的野生鸟类中出现,导致超过7000万只鸟类死亡。当这些毒株传播到鳍足类动物时,也观察到了显著的死亡率。对从鳍足类动物中分离出的HPIAV进行详细的基因特征分析,对于了解这些病毒向包括人类在内的其他哺乳动物物种的潜在传播至关重要。为了深入了解这些问题,对从鳍足类动物中分离出的HPIAV H5N1 2.3.4.4b毒株进行了详细的系统发育分析。这些研究结果揭示了在世界所有地区都存在从鸟类到鳍足类动物的多次传播事件。HPIAV H5N1 2.3.4.4b毒株不同基因的不同进化历史导致了感染世界不同地区鳍足类动物的病毒产生。从鳍足类动物中分离出的欧洲毒株与从南美分离出的毒株代表了完全不同的遗传谱系。从鳍足类动物中分离出的所有毒株都具有对家禽高致病性禽流感的特征。在所有研究的鳍足类物种的不同基因中都观察到了先前显示出对感染哺乳动物具有适应性优势的氨基酸替换。