Zheng XuWen, Chen MaoBing, Zhuang Yi, Zhao Liang, Qian YongJun, Shi ChengCheng
Emergency Department, Wujin People's Hospital Affiliated with Jiangsu University, Wujin Clinical College of Xuzhou Medical University, Changzhou, Jiangsu, China.
Front Microbiol. 2024 Sep 20;15:1448629. doi: 10.3389/fmicb.2024.1448629. eCollection 2024.
Multiple studies suggest a potential connection between the gut microbiome and asthma. Our objective is to use advanced genetic and metagenomic techniques to elucidate the causal relationships and underlying mechanisms between gut microbiota and asthma.
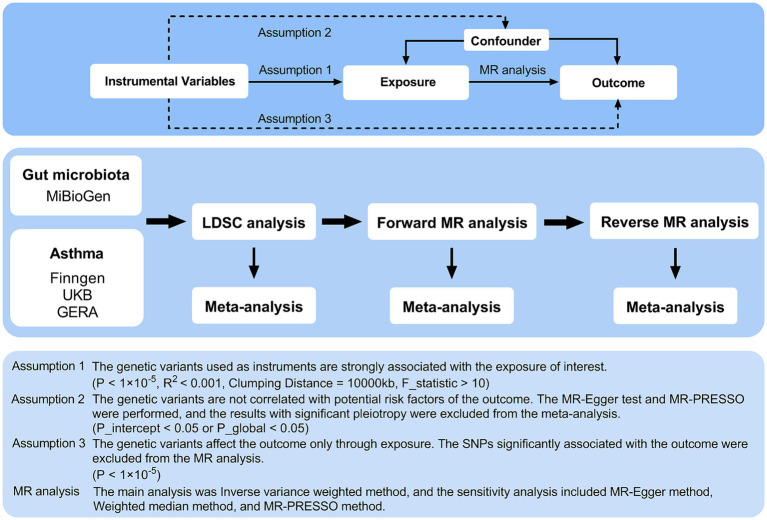
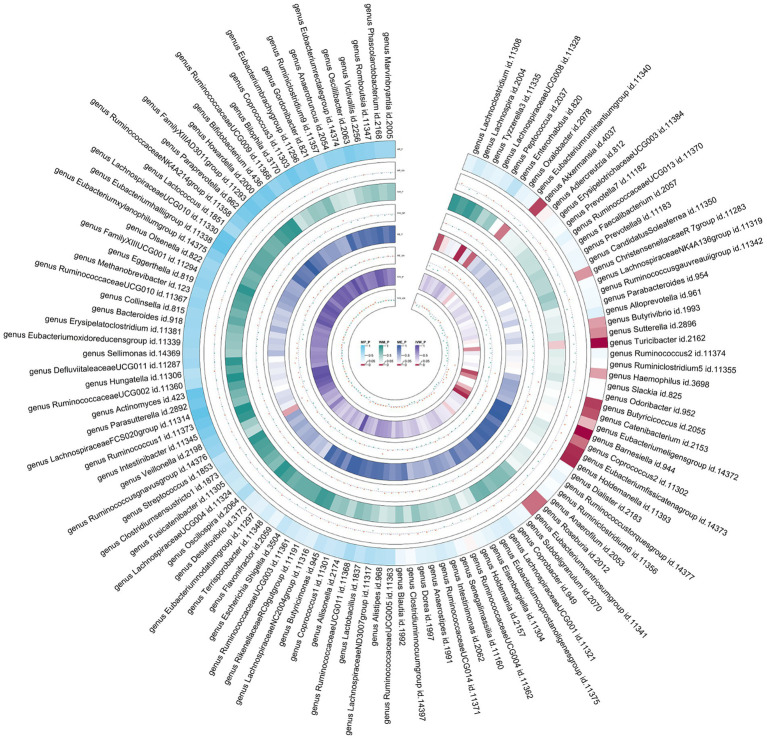
The study utilized comprehensive Linkage Disequilibrium Score Regression (LDSC) and Mendelian randomization (MR) analyses to examine the relationship between 119 gut microbiota genera and asthma, using publicly accessible genome-wide association studies (GWAS). The meta-analysis synthesized summary effect estimates obtained from LDSC, forward MR, and reverse MR. The MiBioGen collaboration, involving 18,340 individuals, identified genetic variations associated with gut bacteria. Asthma data were collected from the UK Biobank, FinnGen, and GERA, encompassing a total of 82,060 cases and 641,049 controls.
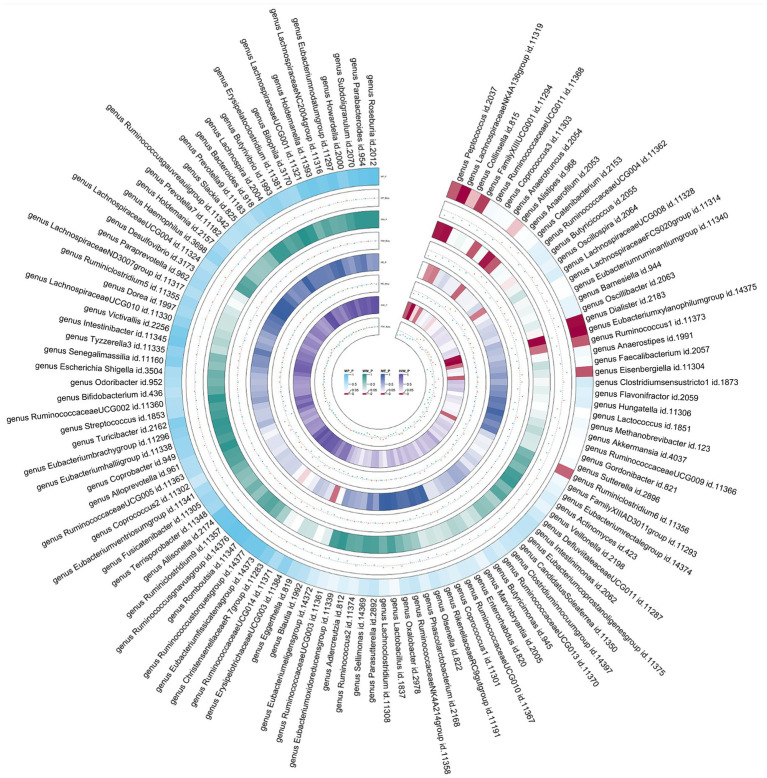
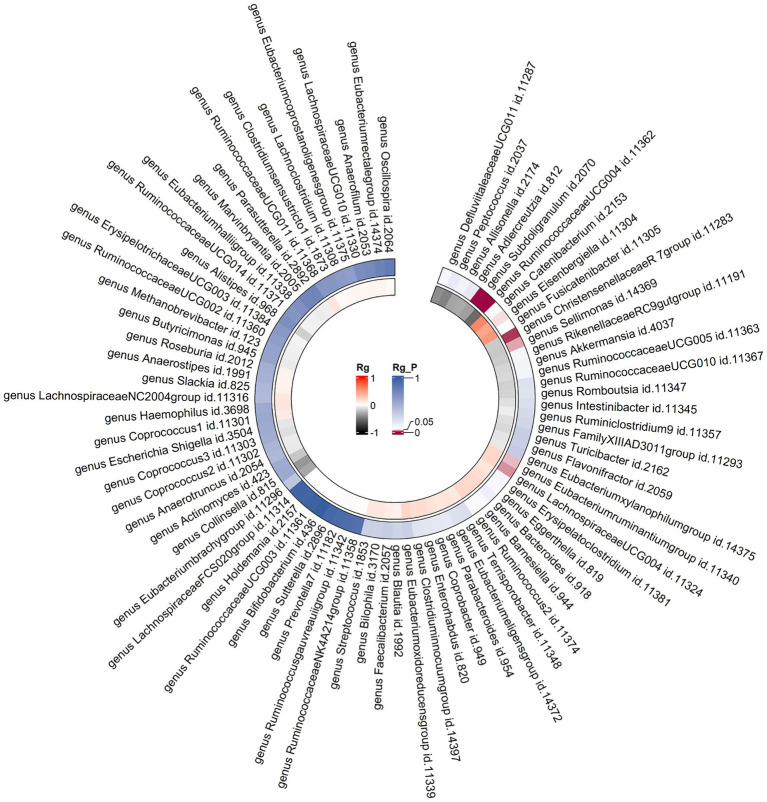
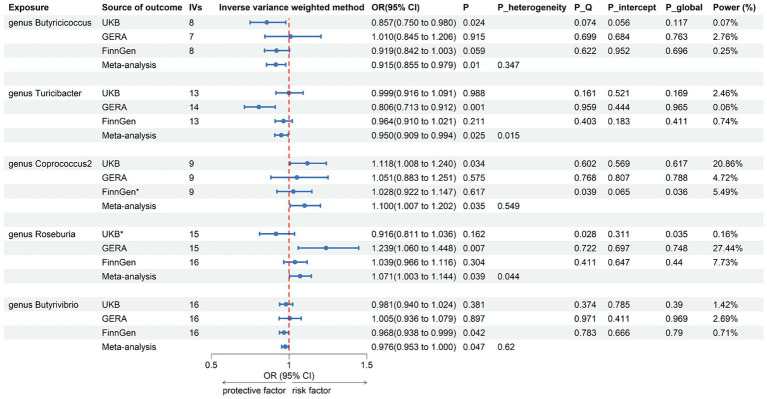
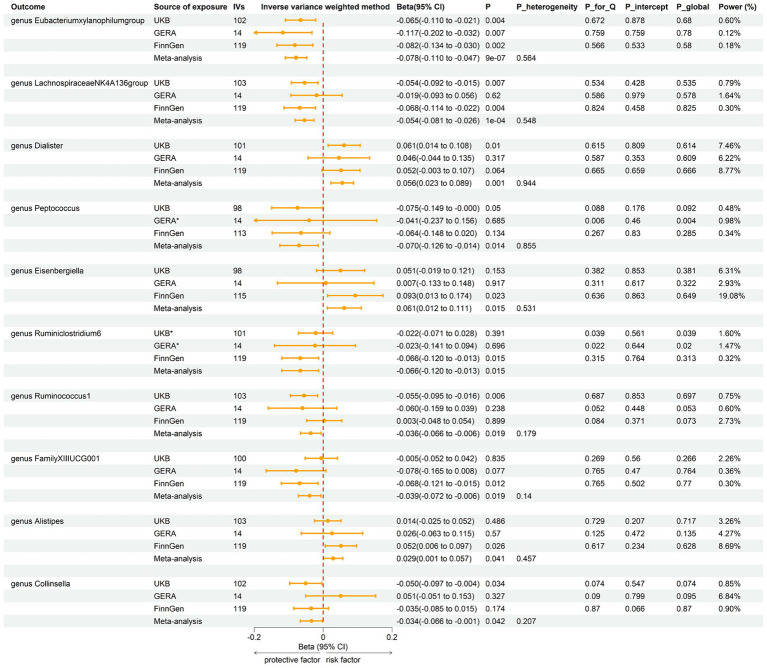
LDSC analysis revealed significant negative genetic correlations between asthma and (Rg = -0.55, = 7.66 × 10) and (Rg = -0.35, = 3.61 × 10). Forward MR analysis suggested associations between (OR = 0.92, = 0.01), (OR = 0.95, = 0.025), (OR = 0.98, = 0.047), and reduced asthma risk. Conversely, (OR = 1.10, = 0.035) and (OR = 1.07, = 0.039) were associated with increased risk. Reverse MR analysis indicated significant associations between genetically predicted asthma and (Beta = -0.08, = 9.25 × 10), (Beta = -0.05, = 1.26 × 10), and (Beta = 0.06, = 0.015, Rg_ = 0.043).
The findings underscore significant genetic correlations and causal relationships between specific gut microbiota and asthma. These insights highlight the potential of gut microbiota as both markers and modulators of asthma risk, offering new avenues for targeted therapeutic strategies.
多项研究表明肠道微生物群与哮喘之间存在潜在联系。我们的目标是使用先进的基因和宏基因组技术来阐明肠道微生物群与哮喘之间的因果关系及潜在机制。
该研究利用综合连锁不平衡评分回归(LDSC)和孟德尔随机化(MR)分析,通过公开可用的全基因组关联研究(GWAS)来检验119种肠道微生物属与哮喘之间的关系。荟萃分析综合了从LDSC、正向MR和反向MR获得的汇总效应估计值。MiBioGen合作项目涉及18340名个体,确定了与肠道细菌相关的基因变异。哮喘数据收集自英国生物银行、芬兰基因研究项目和基因与环境相关性研究(GERA),总共包括82060例病例和641049名对照。
LDSC分析显示哮喘与[具体微生物属1](Rg = -0.55,P = 7.66×10)和[具体微生物属2](Rg = -0.35,P = 3.61×10)之间存在显著的负遗传相关性。正向MR分析表明[具体微生物属3](OR = 0.92,P = 0.01)、[具体微生物属4](OR = 0.95,P = 0.025)、[具体微生物属5](OR = 0.98,P = 0.047)与降低哮喘风险相关。相反,[具体微生物属6](OR = 1.10,P = 0.035)和[具体微生物属7](OR = 1.07,P = 0.039)与风险增加相关。反向MR分析表明基因预测的哮喘与[具体微生物属8](Beta = -0.08,P = 9.25×10)[具体微生物属9](Beta = -0.05,P = 1.26×10)和[具体微生物属10](Beta = 0.06,P = 0.015,Rg_ = 0.043)之间存在显著关联。
这些发现强调了特定肠道微生物群与哮喘之间存在显著的遗传相关性和因果关系。这些见解突出了肠道微生物群作为哮喘风险标志物和调节因子的潜力,为靶向治疗策略提供了新途径。