Flowers Jessica, Echols Nathaniel, Correy Galen, Jaishankar Priya, Togo Takaya, Renslo Adam R, van den Bedem Henry, Fraser James S, Wankowicz Stephanie A
Department of Bioengineering and Therapeutic Sciences, University of California San Francisco, San Francisco, CA.
Department of Pharmaceutical Chemistry, University of California San Francisco, San Francisco, CA.
bioRxiv. 2024 Sep 23:2024.09.20.613996. doi: 10.1101/2024.09.20.613996.
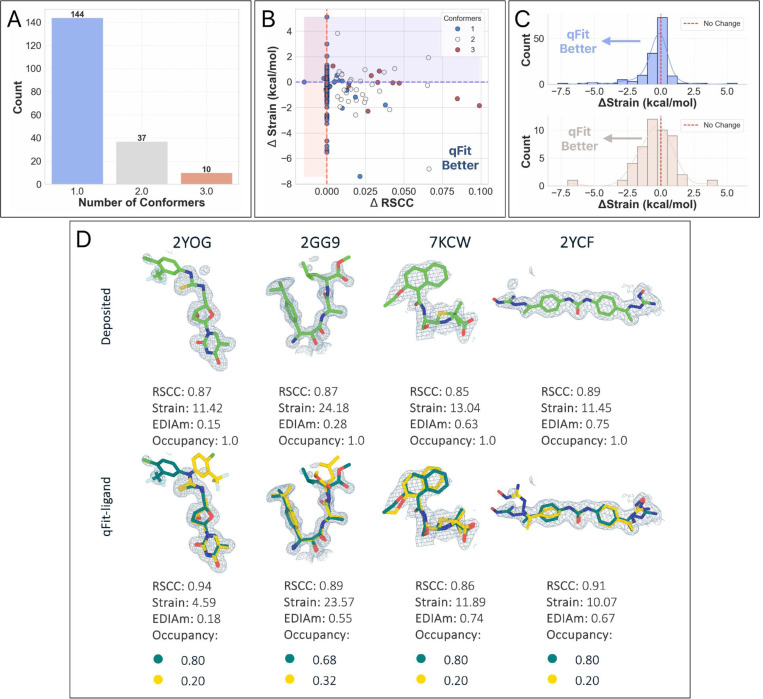
Small molecule ligands exhibit a diverse range of conformations in solution. Upon binding to a target protein, this conformational diversity is generally reduced. However, ligands can retain some degree of conformational flexibility even when bound to a receptor. In the Protein Data Bank (PDB), a small number of ligands have been modeled with distinct alternative conformations that are supported by X-ray crystallography density maps. However, the vast majority of structural models are fit to a single ligand conformation, potentially ignoring the underlying conformational heterogeneity present in the sample. We previously developed qFit-ligand to sample diverse ligand conformations and to select a parsimonious ensemble consistent with the density. While this approach indicated that many ligands populate alternative conformations, limitations in our sampling procedures often resulted in non-physical conformations and could not model complex ligands like macrocycles. Here, we introduce several improvements to qFit-ligand, including the use of routines within RDKit for stochastic conformational sampling. This new sampling method greatly enriches low energy conformations of small molecules and macrocycles. We further extended qFit-ligand to identify alternative conformations in PanDDA-modified density maps from high throughput X-ray fragment screening experiments. The new version of qFit-ligand improves fit to electron density and reduces torsional strain relative to deposited single conformer models and our previous version of qFit-ligand. These advances enhance the analysis of residual conformational heterogeneity present in ligand-bound structures, which can provide important insights for the rational design of therapeutic agents.
小分子配体在溶液中呈现出多种构象。在与靶蛋白结合后,这种构象多样性通常会降低。然而,即使与受体结合,配体仍可保留一定程度的构象灵活性。在蛋白质数据库(PDB)中,少数配体已通过X射线晶体学密度图支持的不同替代构象进行建模。然而,绝大多数结构模型都适合单一配体构象,这可能忽略了样品中潜在的构象异质性。我们之前开发了qFit-ligand来对多种配体构象进行采样,并选择与密度一致的简约集合。虽然这种方法表明许多配体存在替代构象,但我们采样程序的局限性常常导致非物理构象,并且无法对大环等复杂配体进行建模。在此,我们对qFit-ligand进行了多项改进,包括使用RDKit中的例程进行随机构象采样。这种新的采样方法极大地丰富了小分子和大环的低能量构象。我们进一步扩展了qFit-ligand,以在高通量X射线片段筛选实验的PanDDA修改密度图中识别替代构象。相对于已沉积的单一构象模型和我们之前版本的qFit-ligand,新版本的qFit-ligand提高了对电子密度的拟合度并减少了扭转应变。这些进展增强了对配体结合结构中存在的残余构象异质性的分析,这可为治疗药物的合理设计提供重要见解。