Department of Rehabilitation Sciences, Faculty of Health and Social Sciences, The Hong Kong Polytechnic University, Hong Kong SAR, China.
Division of Neurology, Department of Medicine, School of Clinical Medicine, The University of Hong Kong, Hong Kong SAR, China.
Cell Commun Signal. 2024 Oct 10;22(1):485. doi: 10.1186/s12964-024-01844-y.
Stress-induced activation of ERK/Drp1 serves as a checkpoint in the segregation of damaged mitochondria for autophagic clearance (mitophagy). Elevated cytosolic calcium (Ca) activates ERK, which is pivotal to mitophagy initiation. This process is altered in Parkinson's disease (PD) with mutations in leucine-rich repeat kinase 2 (LRRK2), potentially contributing to mitochondrial dysfunction. Pathogenic LRRK2 mutation is linked to dysregulated cellular Ca signaling but the mechanism involved remains unclear.
Mitochondrial damages lead to membrane depolarization. To investigate how LRRK2 mutation impairs cellular response to mitochondrial damages, mitochondrial depolarization was induced by artificial uncoupler (FCCP) in wild-type (WT) and LRRK2 mutant knockin (KI) mouse embryonic fibroblasts (MEFs). The resultant cytosolic Ca flux was assessed using live-cell Ca imaging. The role of mitochondria in FCCP-induced cytosolic Ca surge was confirmed by co-treatment with the mitochondrial sodium-calcium exchanger (NCLX) inhibitor. Cellular mitochondrial quality and function were evaluated by Seahorse™ real-time cell metabolic analysis, flow cytometry, and confocal imaging. Mitochondrial morphology was visualized using transmission electron microscopy (TEM). Activation (phosphorylation) of stress response pathways were assessed by immunoblotting.
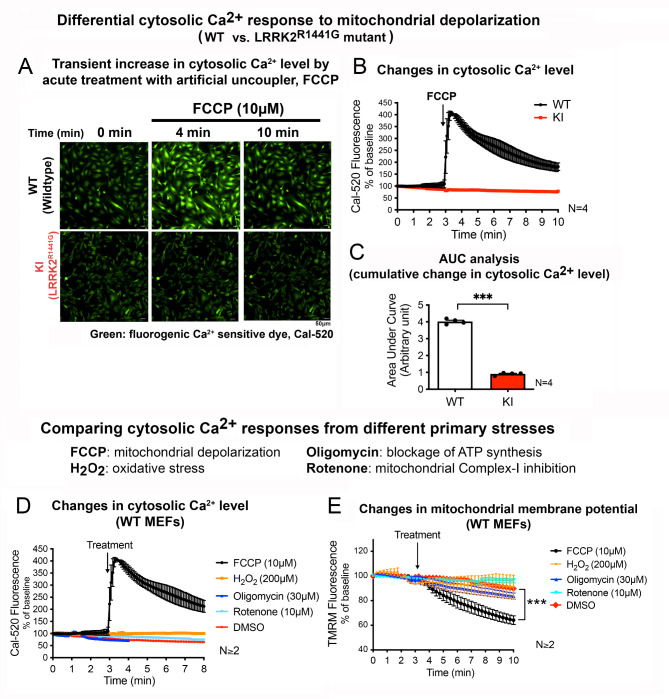
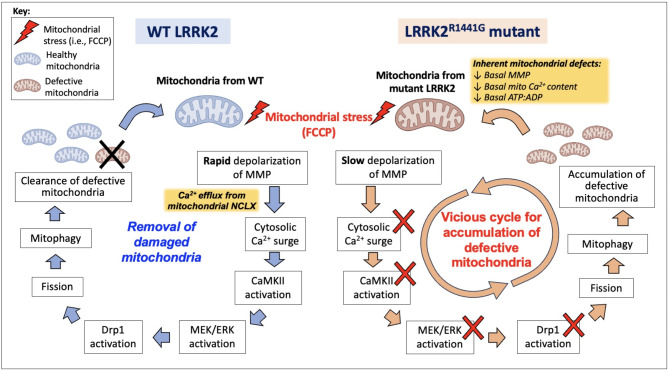
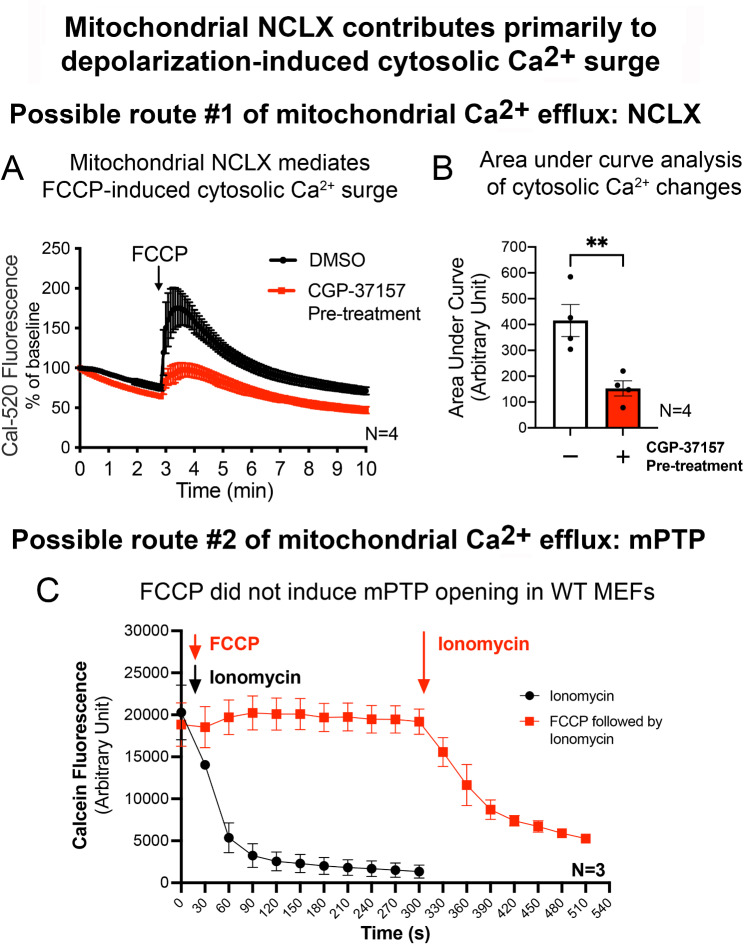
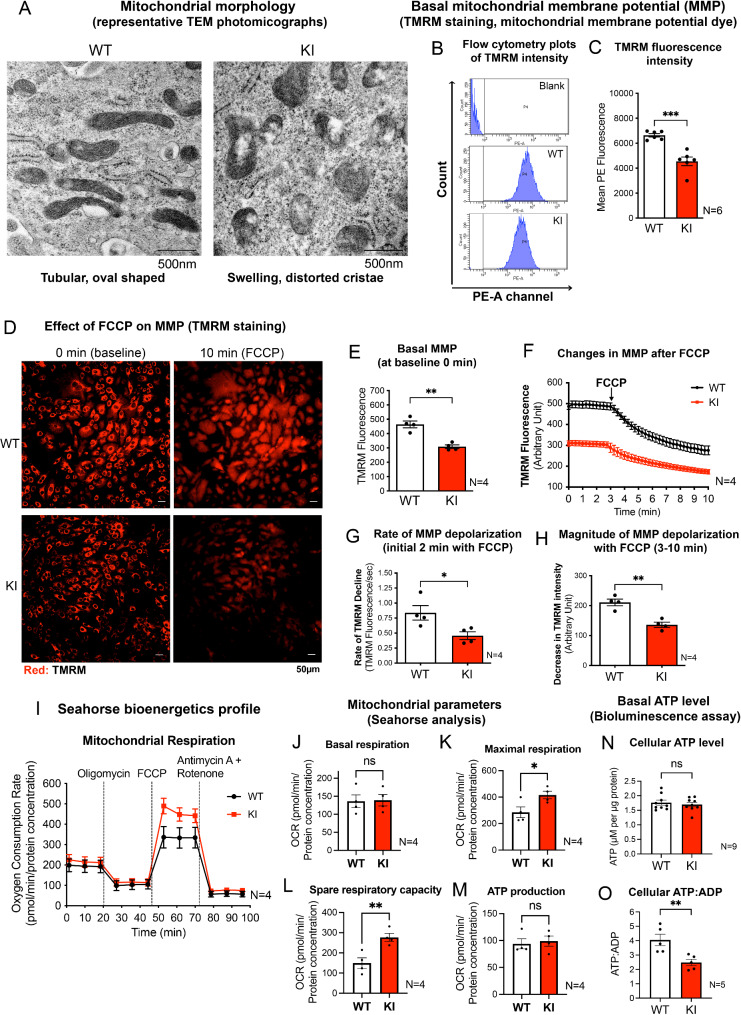
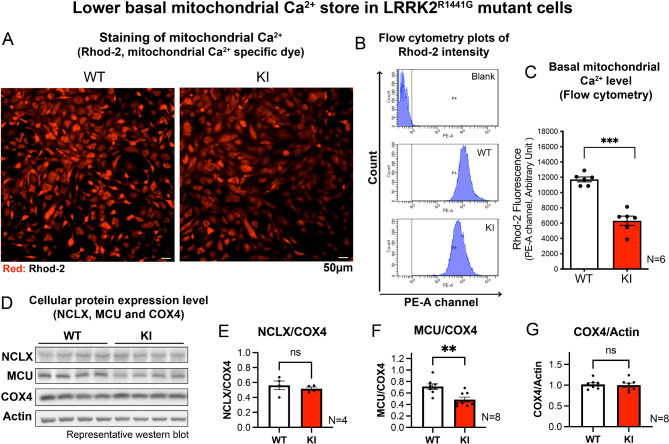
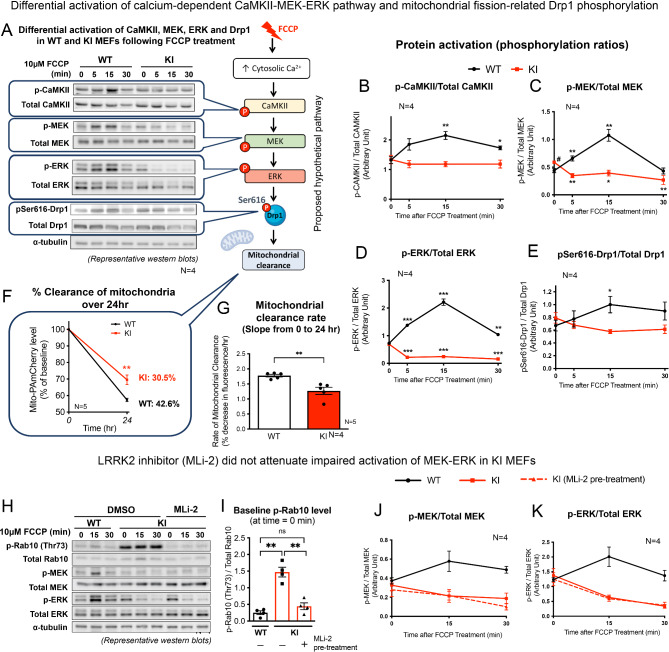
Acute mitochondrial depolarization induced by FCCP resulted in an immediate cytosolic Ca surge in WT MEFs, mediated predominantly via mitochondrial NCLX. However, such cytosolic Ca response was abolished in LRRK2 KI MEFs. This loss of response in KI was associated with impaired activation of Ca/calmodulin-dependent kinase II (CaMKII) and MEK, the two upstream kinases of ERK. Treatment of LRRK2 inhibitor did not rescue this phenotype indicating that it was not caused by mutant LRRK2 kinase hyperactivity. KI MEFs exhibited swollen mitochondria with distorted cristae, depolarized mitochondrial membrane potential, and reduced mitochondrial Ca store and mitochondrial calcium uniporter (MCU) expression. These mutant cells also exhibited lower cellular ATP: ADP ratio albeit higher basal respiration than WT, indicating compensation for mitochondrial dysfunction. These defects may hinder cellular stress response and signals to Drp1-mediated mitophagy, as evident by impaired mitochondrial clearance in the mutant.
Pathogenic LRRK2 mutation abolished mitochondrial depolarization-induced Ca response and impaired the basal mitochondrial clearance. Inherent defects from LRRK2 mutation have weakened the cellular ability to scavenge damaged mitochondria, which may further aggravate mitochondrial dysfunction and neurodegeneration in PD.
应激诱导的 ERK/Drp1 激活可作为受损线粒体分离用于自噬清除(mitophagy)的检查点。胞质 Ca 升高(Ca)激活 ERK,这对于启动 mitophagy 至关重要。帕金森病(PD)中富含亮氨酸重复激酶 2(LRRK2)的突变改变了这一过程,可能导致线粒体功能障碍。致病性 LRRK2 突变与细胞内 Ca 信号的失调有关,但涉及的机制仍不清楚。
线粒体损伤导致膜去极化。为了研究 LRRK2 突变如何损害细胞对线粒体损伤的反应,我们在野生型(WT)和 LRRK2 突变敲入(KI)小鼠胚胎成纤维细胞(MEFs)中使用人工解偶联剂(FCCP)诱导线粒体去极化。使用活细胞 Ca 成像评估由此产生的胞质 Ca 流。通过与线粒体钠钙交换器(NCLX)抑制剂共同处理,证实了线粒体在 FCCP 诱导的胞质 Ca 激增中的作用。通过 Seahorse™实时细胞代谢分析、流式细胞术和共聚焦成像评估细胞线粒体质量和功能。使用透射电子显微镜(TEM)观察线粒体形态。通过免疫印迹评估应激反应途径的激活(磷酸化)。
FCCP 诱导的急性线粒体去极化导致 WT MEFs 中立即发生胞质 Ca 激增,主要通过线粒体 NCLX 介导。然而,KI MEFs 中这种胞质 Ca 反应被消除。KI 中这种反应的丧失与 Ca/钙调蛋白依赖性激酶 II(CaMKII)和 MEK 的激活受损有关,CaMKII 和 MEK 是 ERK 的两个上游激酶。用 LRRK2 抑制剂处理不能挽救这种表型,表明这不是由于突变 LRRK2 激酶过度活跃引起的。KI MEFs 显示出线粒体肿胀,嵴扭曲,线粒体膜电位去极化,线粒体 Ca 库和线粒体钙单向转运体(MCU)表达减少。这些突变细胞还表现出较低的细胞 ATP:ADP 比值,尽管基础呼吸率较高,表明对线粒体功能障碍的补偿。这些缺陷可能会阻碍细胞应激反应和 Drp1 介导的 mitophagy 的信号,这从突变体中线粒体清除受损中可以明显看出。
致病性 LRRK2 突变消除了线粒体去极化诱导的 Ca 反应,并损害了基础线粒体清除。LRRK2 突变固有的缺陷削弱了细胞清除受损线粒体的能力,这可能进一步加重 PD 中的线粒体功能障碍和神经退行性变。