Department of Infectious Diseases, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, 450052, Henan, China.
Department of Oncology, The First Affiliated Hospital, College of Clinical Medicine, Henan University of Science and Technology, Luoyang, 471000, Henan, China.
Sci Rep. 2024 Oct 11;14(1):23832. doi: 10.1038/s41598-024-75609-5.
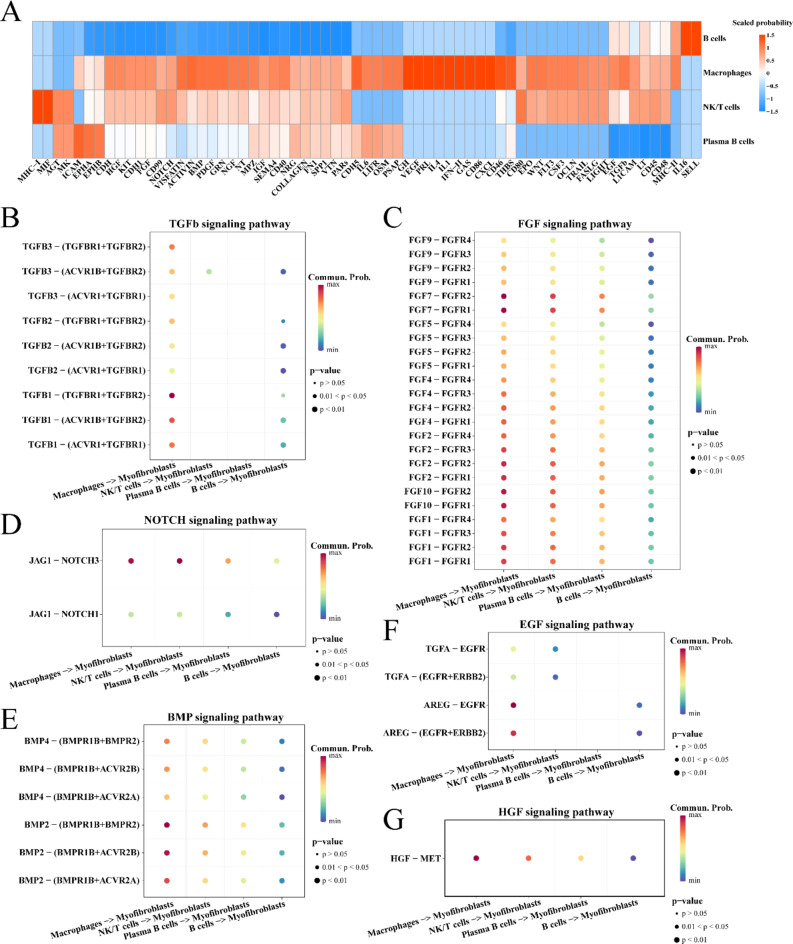
Hepatocellular carcinoma with cirrhosis promotes the advancement of malignancy and the development of fibrosis in normal liver tissues. Understanding the pathological mechanisms underlying the development of HCC with cirrhosis is important for developing effective therapeutic strategies. Herein, the RNA-sequencing (RNA-seq) data and corresponding clinical features of patients with HCC were extracted from The Cancer Genome Atlas (TCGA) database using the University of California Santa Cruz (UCSC) Xena platform. The enrichment degree of hallmarkers for each TCGA-LIHC cohort was quantified by ssGSEA algorithm. Weighted gene co-expression network analysis (WGCNA) revealed two gene module eigengenes (MEs) associated with cirrhosis, namely, MEbrown and MEgreen. Analysis of these modules using AUCell showed that MEbrown had higher enrichment scores in all immune cells, whereas MEgreen had higher enrichment scores in malignant cells. The CellChat package revealed that both immune and malignant cells contributed to the fibrotic activity of myofibroblasts through diverse signaling pathways. Additionally, spatial transcriptomic data showed that hepatocytes, proliferating hepatocytes, macrophages, and myofibroblasts were located in closer proximity in HCC tissues. These cells may potentially participate in the process of stimulating myofibroblast fibrotic activity, which may be related to the development of liver fibrosis. In summary, we made full use of multi-omics data to explore gene networks and cell types that may be involved in the development and progression of cirrhosis in HCC.
伴有肝硬化的肝细胞癌促进了恶性肿瘤的进展和正常肝组织的纤维化发展。了解伴有肝硬化的 HCC 发展的病理机制对于开发有效的治疗策略非常重要。在此,使用加利福尼亚大学圣克鲁兹分校(UCSC)Xena 平台从癌症基因组图谱(TCGA)数据库中提取了 HCC 患者的 RNA 测序(RNA-seq)数据和相应的临床特征。通过 ssGSEA 算法量化了每个 TCGA-LIHC 队列 hallmarker 的富集程度。加权基因共表达网络分析(WGCNA)揭示了与肝硬化相关的两个基因模块特征基因(ME),即 MEbrown 和 MEgreen。使用 AUCell 对这些模块进行分析表明,MEbrown 在所有免疫细胞中的富集评分更高,而 MEgreen 在恶性细胞中的富集评分更高。CellChat 包表明,免疫细胞和恶性细胞均通过多种信号通路促进成纤维细胞肌成纤维细胞的纤维化活性。此外,空间转录组学数据表明,肝癌组织中的肝细胞、增殖肝细胞、巨噬细胞和成纤维细胞彼此更接近。这些细胞可能潜在地参与刺激成纤维细胞肌成纤维细胞的纤维化活性的过程,这可能与肝纤维化的发展有关。总之,我们充分利用多组学数据来探索可能参与 HCC 中肝硬化发展和进展的基因网络和细胞类型。