Department of Chemotherapy, Guangxi Medical University Cancer Hospital, Nanning, China.
Department of Medical Oncology, Fujian Medical University Union Hospital, Fuzhou, China.
Pathol Oncol Res. 2023 Jan 4;28:1610808. doi: 10.3389/pore.2022.1610808. eCollection 2022.
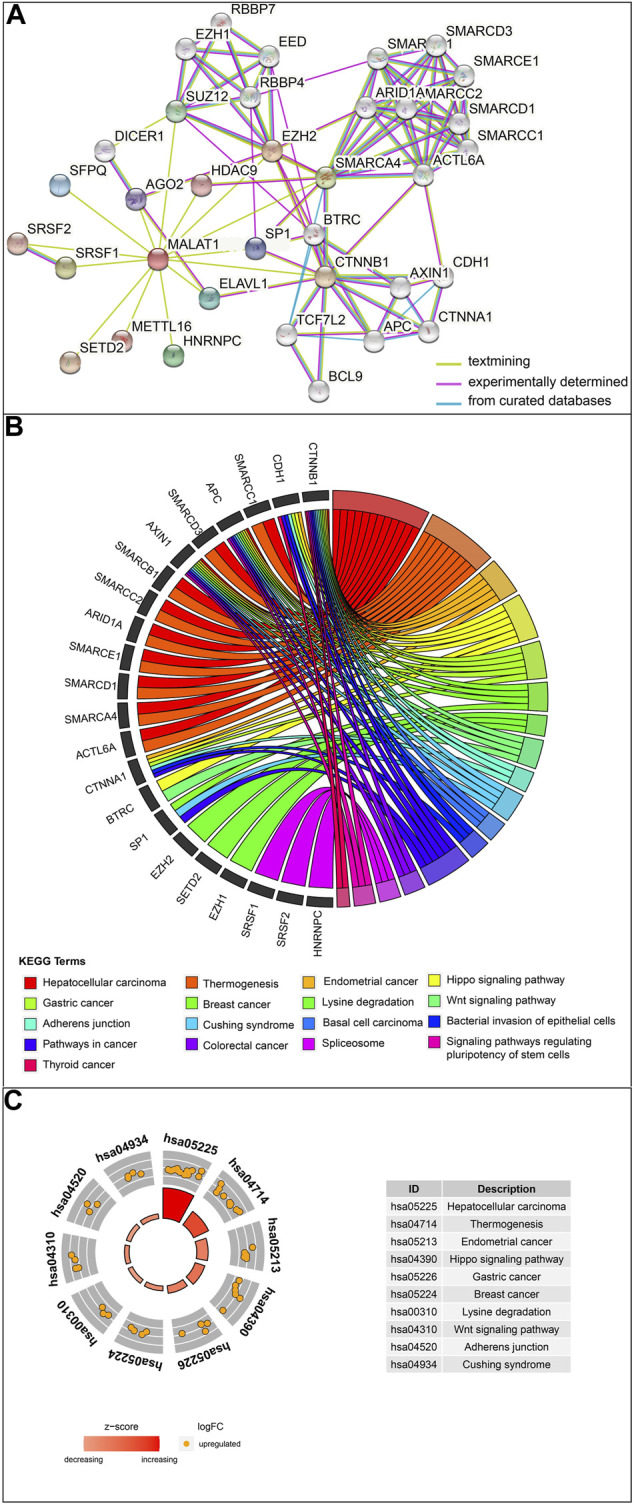
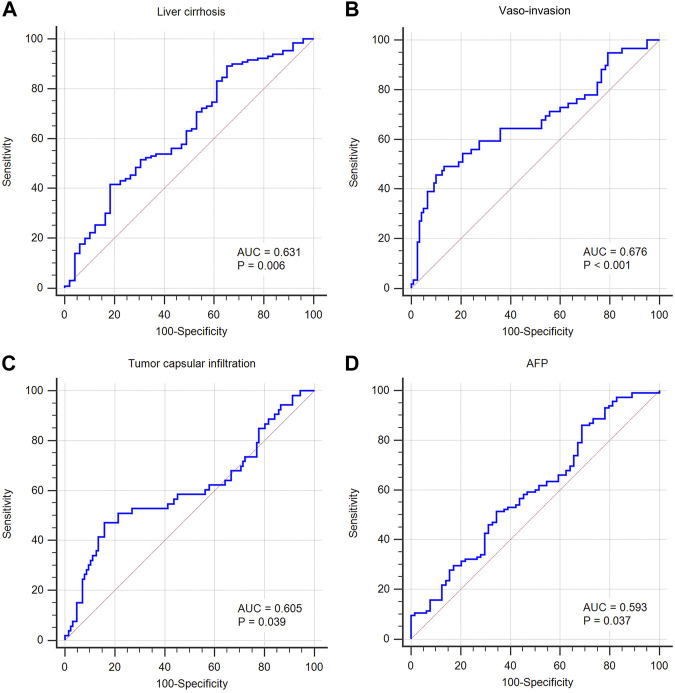
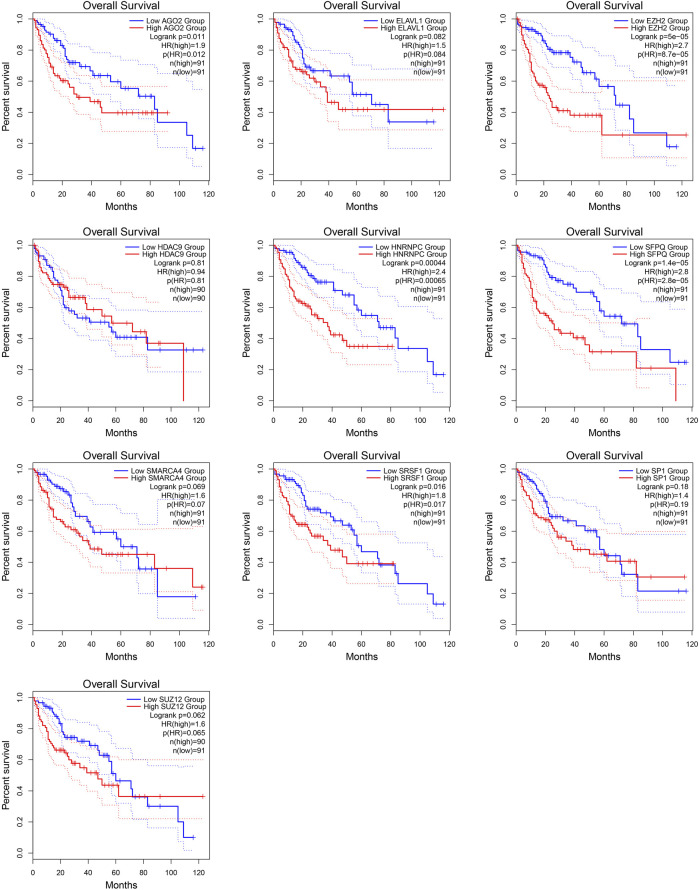
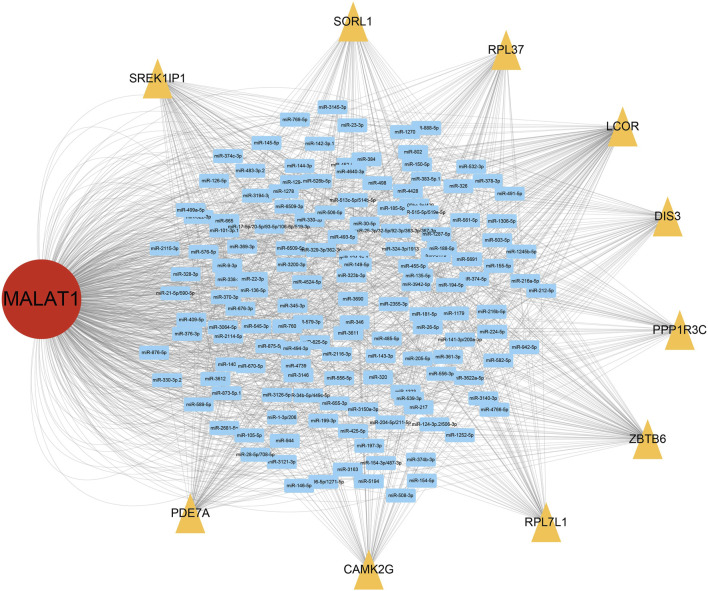
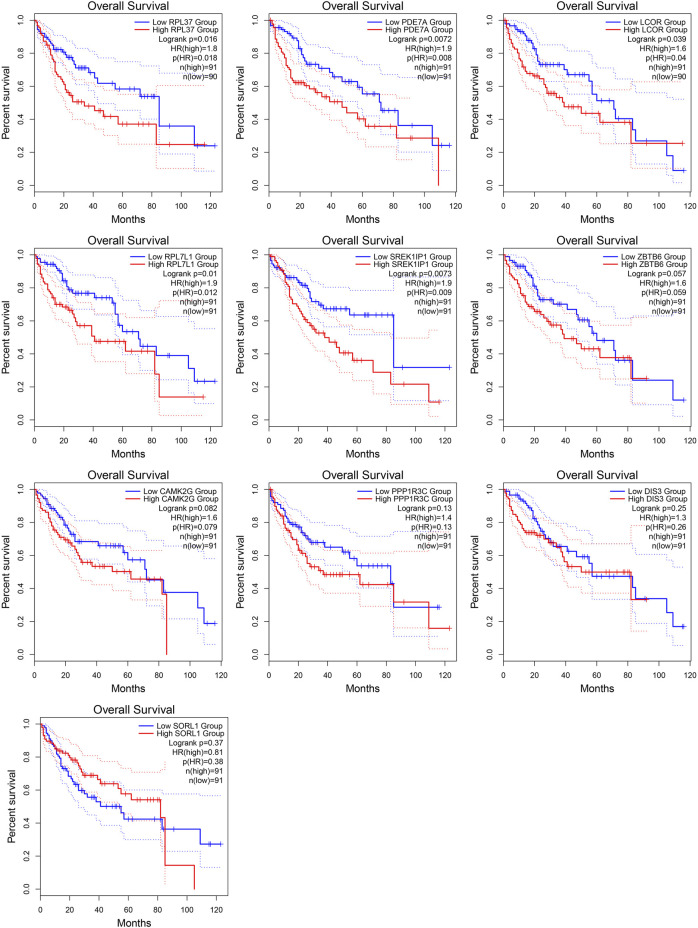
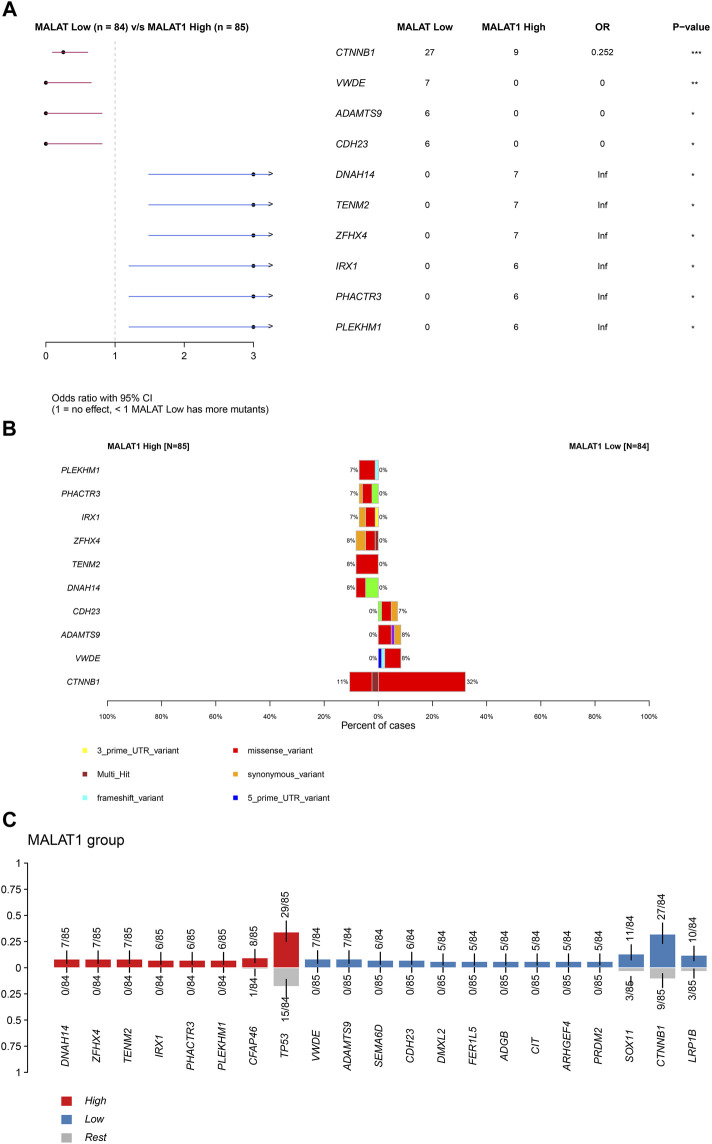
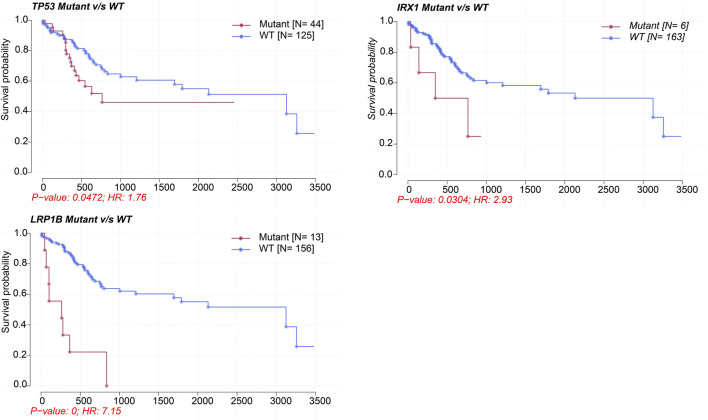
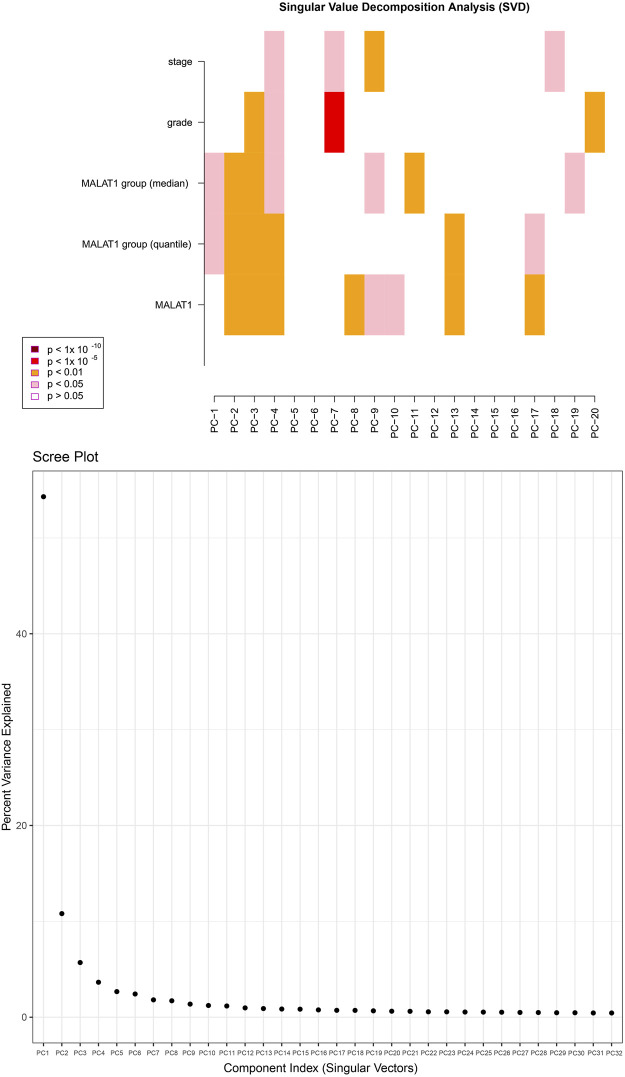
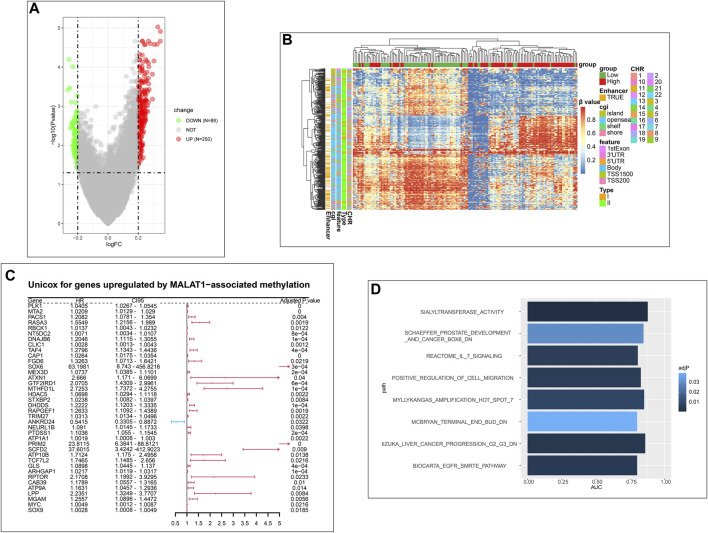
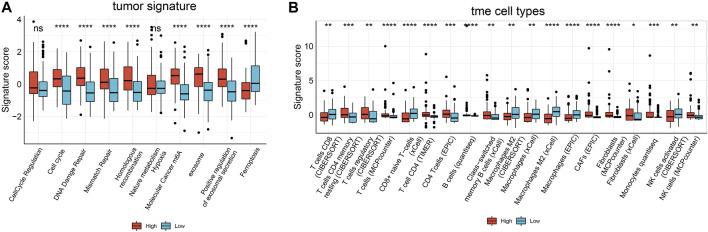
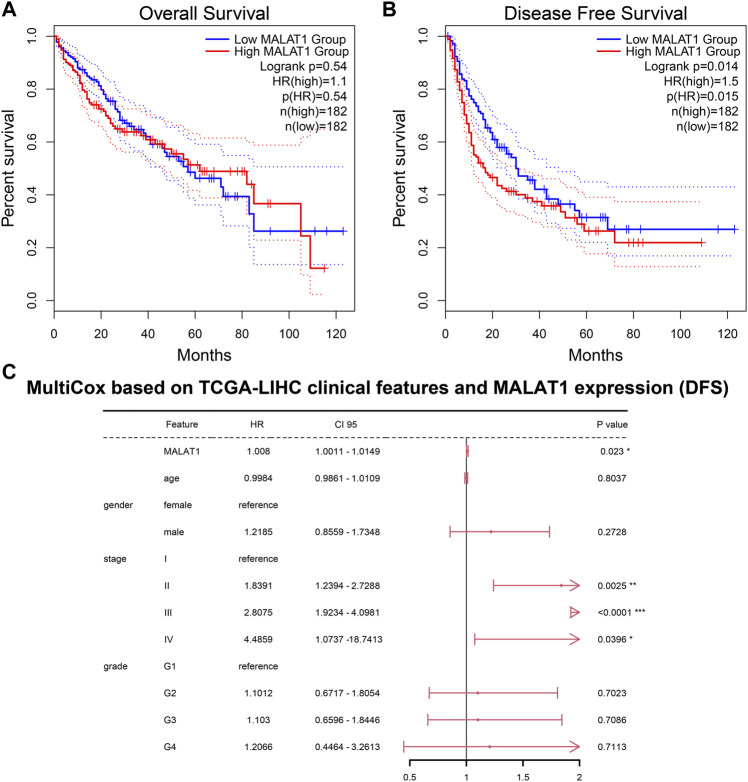
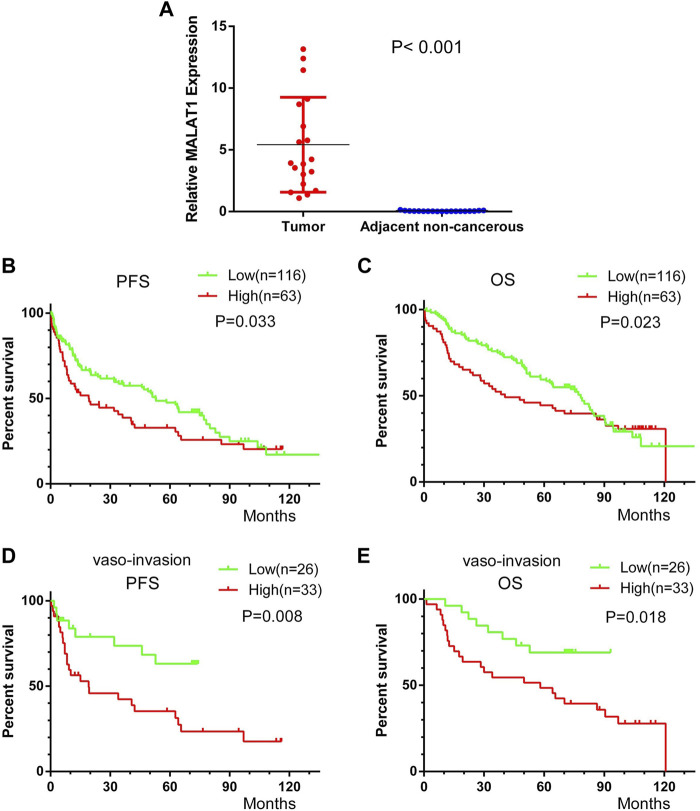
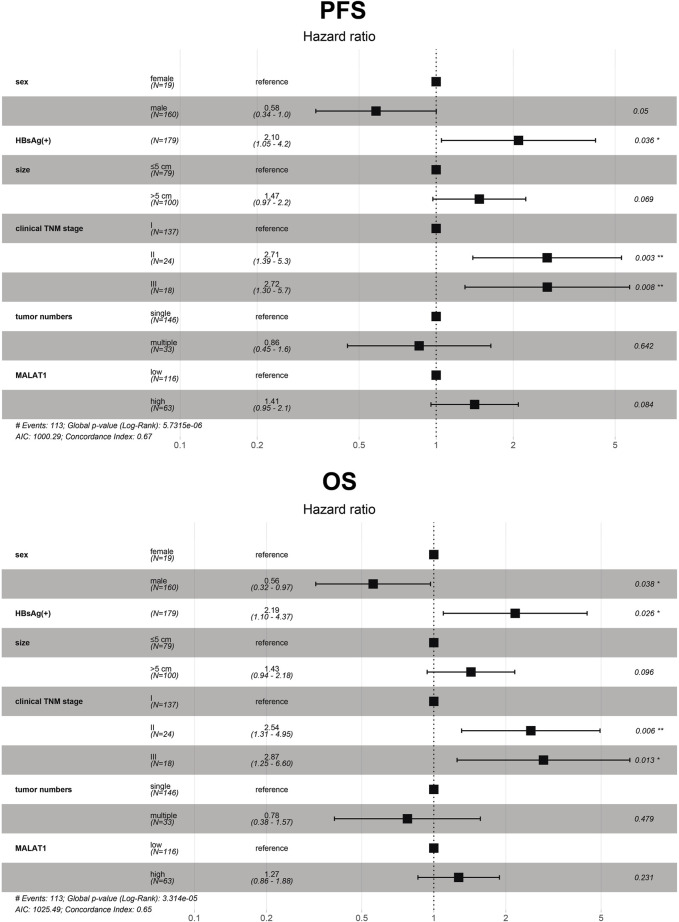
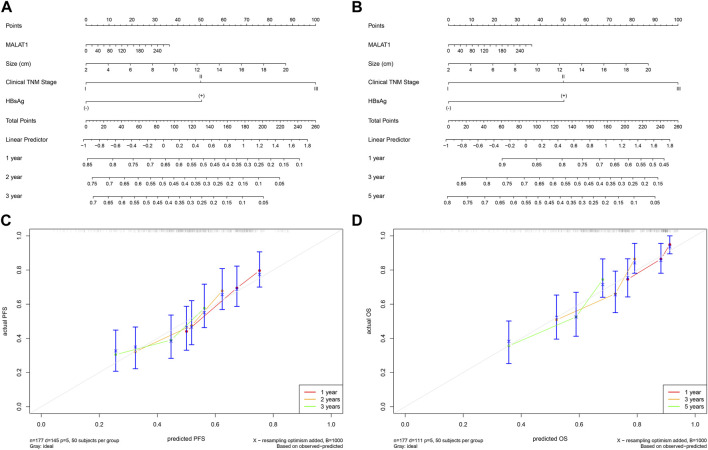
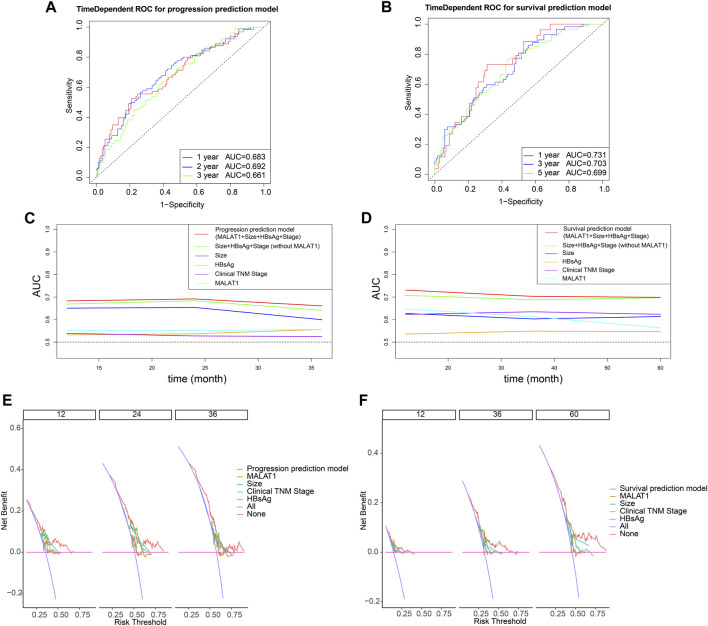
This study aimed to explore the relationship between and the prognosis of patients with hepatocellular carcinoma (HCC). We constructed a protein-protein interaction network using the STRING database and a network of competing endogenous RNAs (ceRNAs) using the StarBase database. Using data from the GEPIA2 database, we studied the association between genes in these networks and survival of patients with HCC. The potential mechanisms underlying the relationship between and HCC prognosis were studied using combined data from RNA sequencing, DNA methylation, and somatic mutation data from The Cancer Genome Atlas (TCGA) liver cancer cohort. Tumor tissues and 19 paired adjacent non-tumor tissues (PANTs) from HCC patients who underwent radical resection were analyzed for mRNA levels using real-time PCR, and associations of expression with clinicopathological features or prognosis of patients were analyzed using log-rank test and Gehan-Breslow-Wilcoxon test. Five interacting proteins and five target genes of in the ceRNA network significantly correlated with poor survival of patients with HCC ( < 0.05). High expression was associated with mutations in two genes leading to poor prognosis and may upregulate some prognostic risk genes through methylation. was significantly co-expressed with various signatures of genes involved in HCC progression, including the cell cycle, DNA damage repair, mismatch repair, homologous recombination, molecular cancer m6A, exosome, ferroptosis, infiltration of lymphocyte ( < 0.05). The expression of was markedly upregulated in HCC tissues compared with PANTs. In Kaplan-Meier analysis, patients with high expression had significantly shorter progression-free survival (PFS) ( = 0.033) and overall survival (OS) ( = 0.023) than those with low expression. Median PFS was 19.2 months for patients with high expression and 52.8 months for patients with low expression, while the corresponding median OS was 40.5 and 78.3 months. In subgroup analysis of patients with vascular invasion, cirrhosis, and HBsAg positive or AFP positive, MALAT1 overexpression was significantly associated with shorter PFS and OS. Models for predicting PFS and OS constructed based on expression and clinicopathological features had moderate predictive power, with areas under the receiver operating characteristic curves of 0.661-0.731. Additionally, expression level was significantly associated with liver cirrhosis, vascular invasion, and tumor capsular infiltration ( < 0.05 for all). is overexpressed in HCC, and higher expression is associated with worse prognosis. mRNA level may serve as a prognostic marker for patients with HCC after hepatectomy.
本研究旨在探讨 与肝细胞癌(HCC)患者预后的关系。我们使用 STRING 数据库构建了一个 蛋白-蛋白相互作用网络,使用 StarBase 数据库构建了一个竞争性内源 RNA(ceRNA)网络。使用 GEPIA2 数据库中的数据,我们研究了这些网络中的基因与 HCC 患者生存之间的关联。使用来自癌症基因组图谱(TCGA)肝癌队列的 RNA 测序、DNA 甲基化和体细胞突变数据的综合数据,研究了 与 HCC 预后之间关系的潜在机制。使用实时 PCR 分析了接受根治性切除术的 HCC 患者的肿瘤组织和 19 对相邻非肿瘤组织(PANT)中的 mRNA 水平,并使用对数秩检验和 Gehan-Breslow-Wilcoxon 检验分析了 表达与患者临床病理特征或预后的关系。ceRNA 网络中与 相互作用的五个蛋白和五个靶基因与 HCC 患者的不良生存显著相关(<0.05)。高 表达与导致预后不良的两个基因的突变相关,并且可能通过甲基化上调一些预后风险基因。与 HCC 进展相关的基因的各种特征明显与 的表达共表达,包括细胞周期、DNA 损伤修复、错配修复、同源重组、分子癌症 m6A、外泌体、铁死亡、淋巴细胞浸润(<0.05)。与 PANT 相比,HCC 组织中 的表达明显上调。在 Kaplan-Meier 分析中,高 表达的患者无进展生存期(PFS)(=0.033)和总生存期(OS)(=0.023)明显短于低表达的患者。高 表达患者的中位 PFS 为 19.2 个月,低表达患者的中位 PFS 为 52.8 个月,相应的中位 OS 分别为 40.5 个月和 78.3 个月。在有血管侵犯、肝硬化、HBsAg 阳性或 AFP 阳性的患者亚组分析中,MALAT1 过表达与较短的 PFS 和 OS 显著相关。基于 表达和临床病理特征构建的预测 PFS 和 OS 的模型具有中等预测能力,受试者工作特征曲线下面积为 0.661-0.731。此外, 的表达水平与肝硬化、血管侵犯和肿瘤包膜浸润显著相关(均<0.05)。 在 HCC 中过表达,高表达与预后不良相关。MALAT1 mRNA 水平可能可作为 HCC 患者肝切除术后的预后标志物。