Material Measurement Laboratory, National Institute of Standards and Technology, Gaithersburg, MD, USA.
Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX, USA.
Commun Biol. 2024 Oct 13;7(1):1316. doi: 10.1038/s42003-024-06981-1.
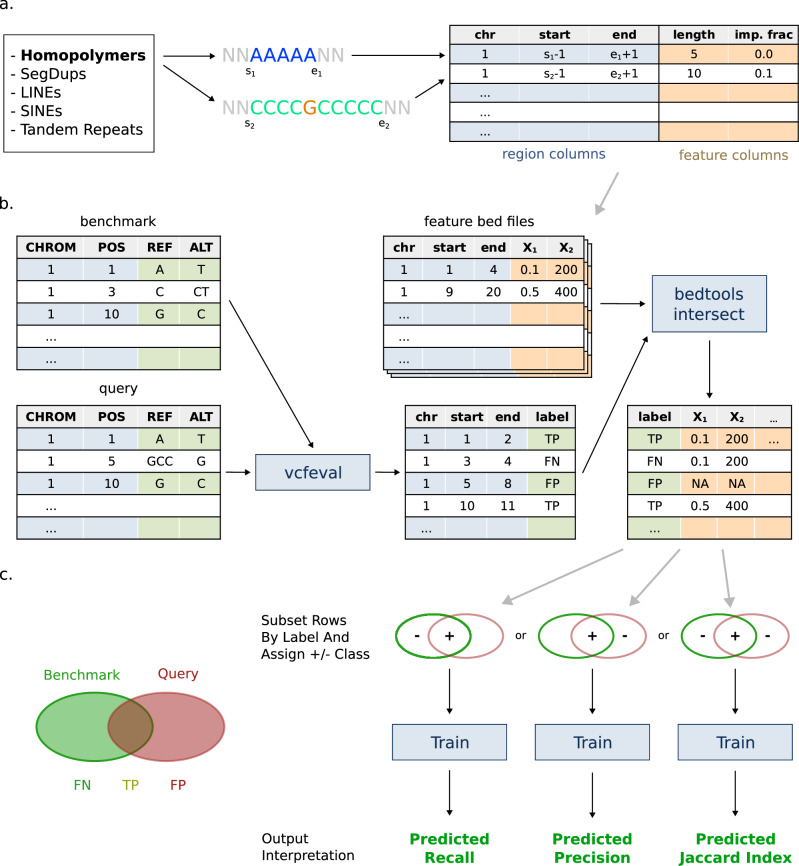
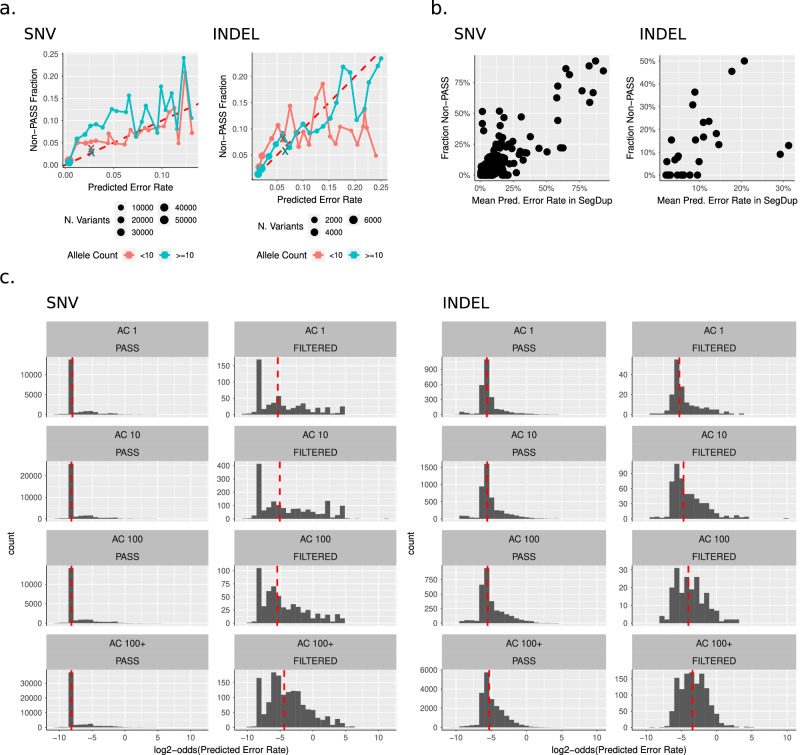
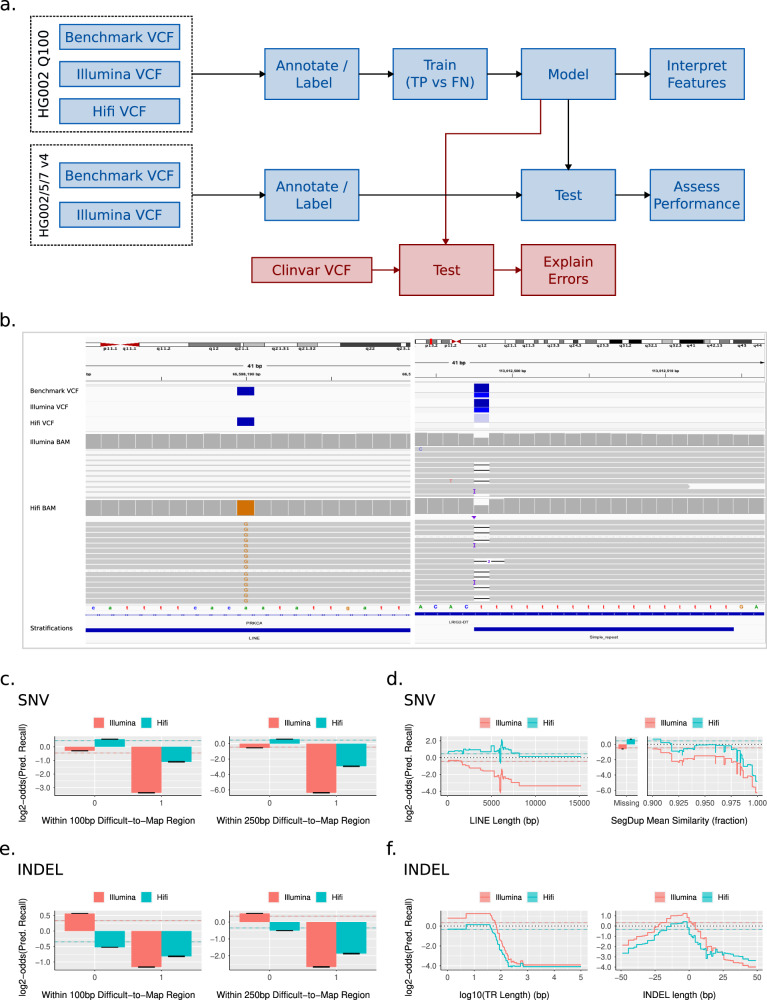
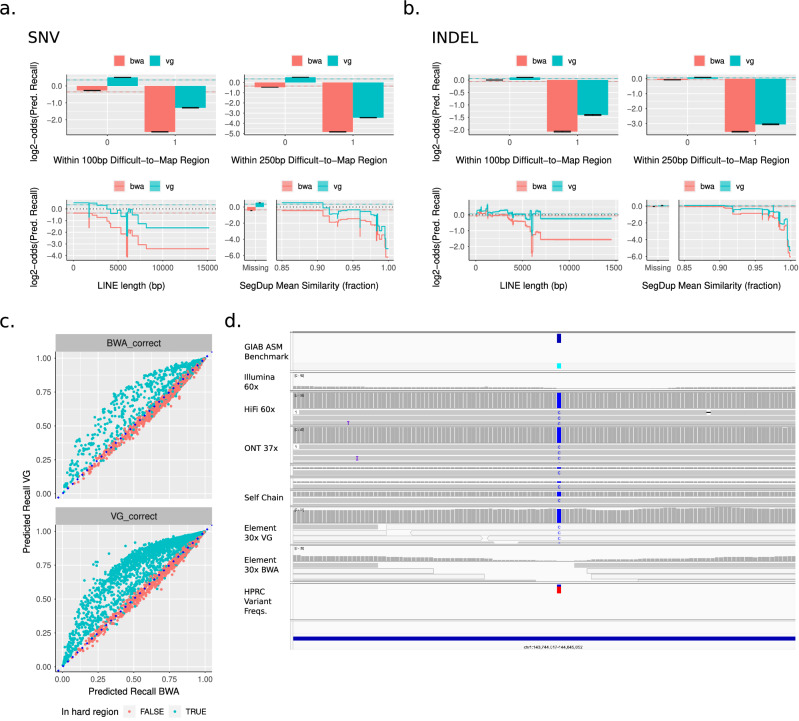
Despite the variety in sequencing platforms, mappers, and variant callers, no single pipeline is optimal across the entire human genome. Therefore, developers, clinicians, and researchers need to make tradeoffs when designing pipelines for their application. Currently, assessing such tradeoffs relies on intuition about how a certain pipeline will perform in a given genomic context. We present StratoMod, which addresses this problem using an interpretable machine-learning classifier to predict germline variant calling errors in a data-driven manner. We show StratoMod can precisely predict recall using Hifi or Illumina and leverage StratoMod's interpretability to measure contributions from difficult-to-map and homopolymer regions for each respective outcome. Furthermore, we use Statomod to assess the effect of mismapping on predicted recall using linear vs. graph-based references, and identify the hard-to-map regions where graph-based methods excelled and by how much. For these we utilize our draft benchmark based on the Q100 HG002 assembly, which contains previously-inaccessible difficult regions. Furthermore, StratoMod presents a new method of predicting clinically relevant variants likely to be missed, which is an improvement over current pipelines which only filter variants likely to be false. We anticipate this being useful for performing precise risk-reward analyses when designing variant calling pipelines.
尽管测序平台、映射器和变异呼叫器种类繁多,但没有一个单一的管道在整个人类基因组中都是最优的。因此,开发人员、临床医生和研究人员在为其应用设计管道时需要做出权衡。目前,评估这些权衡依赖于对特定管道在给定基因组背景下的性能的直觉。我们提出了 StratoMod,它使用可解释的机器学习分类器来以数据驱动的方式预测种系变异呼叫错误。我们表明,StratoMod 可以使用 Hifi 或 Illumina 精确地预测召回率,并利用 StratoMod 的可解释性来衡量每个结果中难以映射和同源多聚体区域的贡献。此外,我们使用 Statomod 来评估使用线性与基于图的参考进行错配预测对召回率的影响,并确定基于图的方法表现出色的困难映射区域以及出色的程度。对于这些,我们利用基于 Q100 HG002 组装的我们的草案基准,其中包含以前无法访问的困难区域。此外,StratoMod 提出了一种新的方法来预测可能被遗漏的临床相关变异,这比目前仅过滤可能错误的变异的管道有所改进。我们预计这对于在设计变异呼叫管道时进行精确的风险回报分析将非常有用。