Hull Alexander J, Atilano Magda L, Hallqvist Jenny, Heywood Wendy, Kinghorn Kerri J
Institute of Healthy Ageing, Department of Genetics, Evolution and Environment, University College London, Darwin Building, Gower Street, London, WC1E 6BT, United Kingdom.
Great Ormond Street Institute of Child Health, University College London, 30 Guildford Street, London, WC1N 1EN, United Kingdom.
Hum Mol Genet. 2024 Dec 6;33(24):2111-2122. doi: 10.1093/hmg/ddae143.
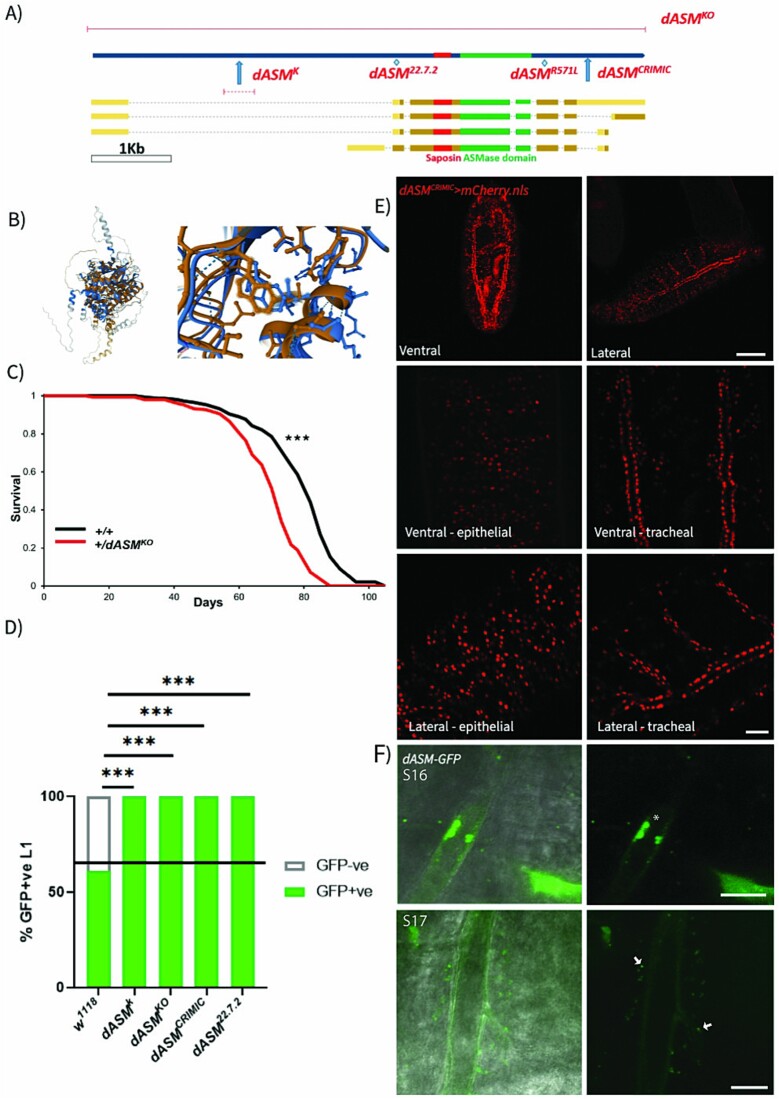
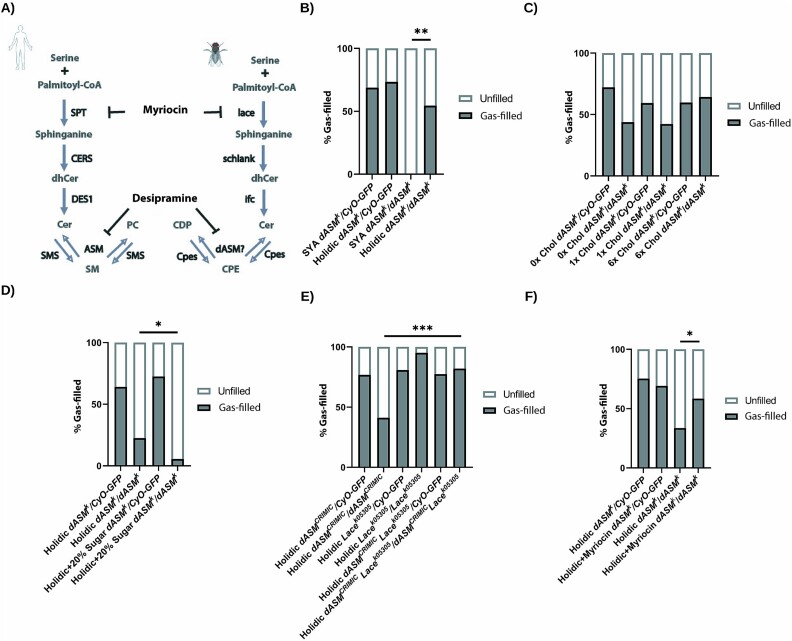
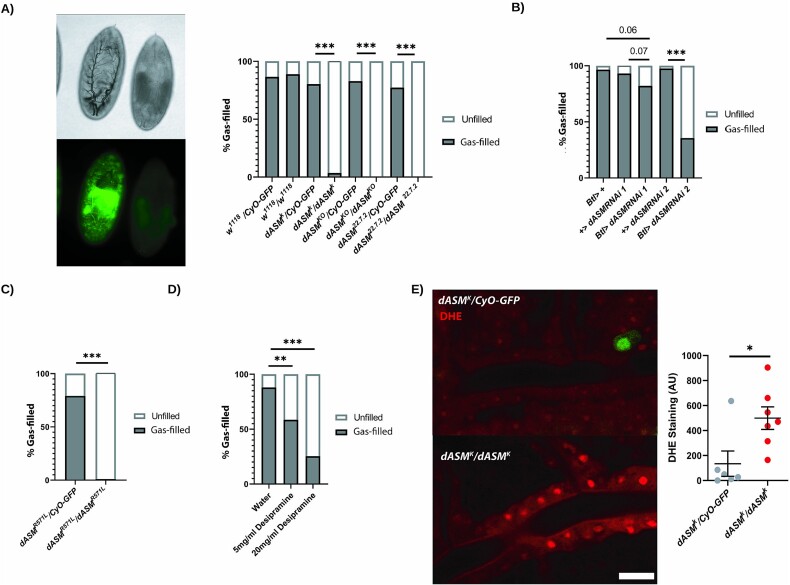
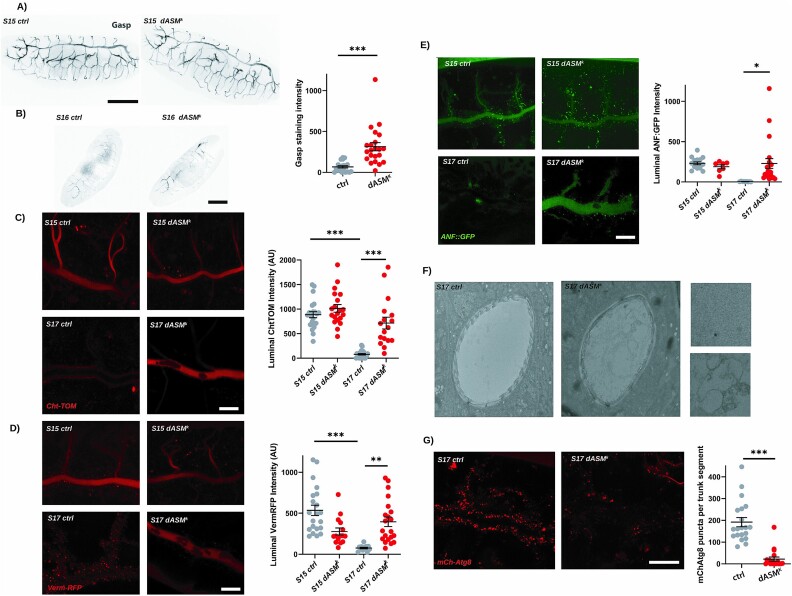
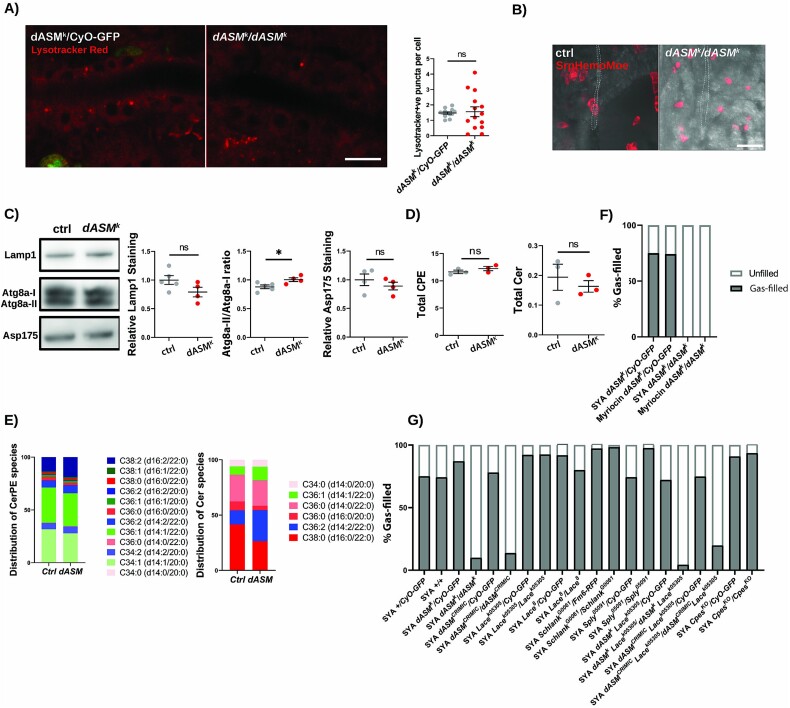
Types A and B Niemann-Pick disease (NPD) are inherited multisystem lysosomal storage disorders due to mutations in the SMPD1 gene. Respiratory dysfunction is a key hallmark of NPD, yet the mechanism for this is underexplored. SMPD1 encodes acid sphingomyelinase (ASM), which hydrolyses sphingomyelin to ceramide and phosphocholine. Here, we present a Drosophila model of ASM loss-of-function, lacking the fly orthologue of SMPD1, dASM, modelling several aspects of the respiratory pathology of NPD. dASM is expressed in the late-embryonic fly respiratory network, the trachea, and is secreted into the tracheal lumen. Loss of dASM results in embryonic lethality, and the tracheal lumen fails to fill normally with gas prior to eclosion. We demonstrate that the endocytic clearance of luminal constituents prior to gas-filling is defective in dASM mutants, and is coincident with autophagic, but not lysosomal defects, in late stage embryonic trachea. Finally, we show that although bulk sphingolipids are unchanged, dietary loss of lipids in combination with genetic and pharmacological block of ceramide synthesis rescues the airway gas-filling defects. We highlight myriocin as a potential therapeutic drug for the treatment of the developmental respiratory defects associated with ASM deficiency, and present a new NPD model amenable to genetic and pharmacological screens.
A型和B型尼曼-匹克病(NPD)是由于SMPD1基因突变导致的遗传性多系统溶酶体贮积症。呼吸功能障碍是NPD的一个关键特征,但其机制尚未得到充分研究。SMPD1编码酸性鞘磷脂酶(ASM),该酶将鞘磷脂水解为神经酰胺和磷酸胆碱。在此,我们展示了一个ASM功能丧失的果蝇模型,该模型缺乏SMPD1的果蝇同源物dASM,模拟了NPD呼吸病理学的几个方面。dASM在胚胎晚期果蝇呼吸网络即气管中表达,并分泌到气管腔中。dASM的缺失导致胚胎致死,并且在羽化前气管腔不能正常充满气体。我们证明,在气体填充之前,管腔成分的内吞清除在dASM突变体中存在缺陷,并且与晚期胚胎气管中的自噬缺陷而非溶酶体缺陷一致。最后,我们表明,尽管总鞘脂类物质没有变化,但饮食中脂质的缺失与神经酰胺合成的遗传和药理学阻断相结合可挽救气道气体填充缺陷。我们强调米里新作为一种潜在的治疗药物,可用于治疗与ASM缺乏相关的发育性呼吸缺陷,并提出了一种适用于遗传和药理学筛选的新的NPD模型。