Abbasabadi Hossein, Bakhtiarizadeh Mohammad Reza, Moradi Mohammad Hossein, McEwan John C
Department of Animal and Poultry Science, College of Aburaihan, University of Tehran, Tehran, Iran.
Department of Animal Sciences, Faculty of Agriculture and Natural Resources, Arak University, Arak, Iran.
Front Vet Sci. 2024 Sep 30;11:1415027. doi: 10.3389/fvets.2024.1415027. eCollection 2024.
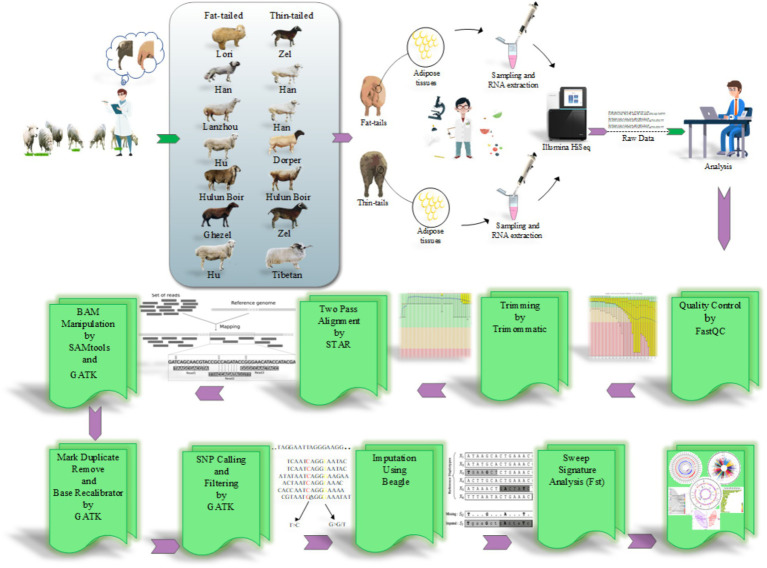
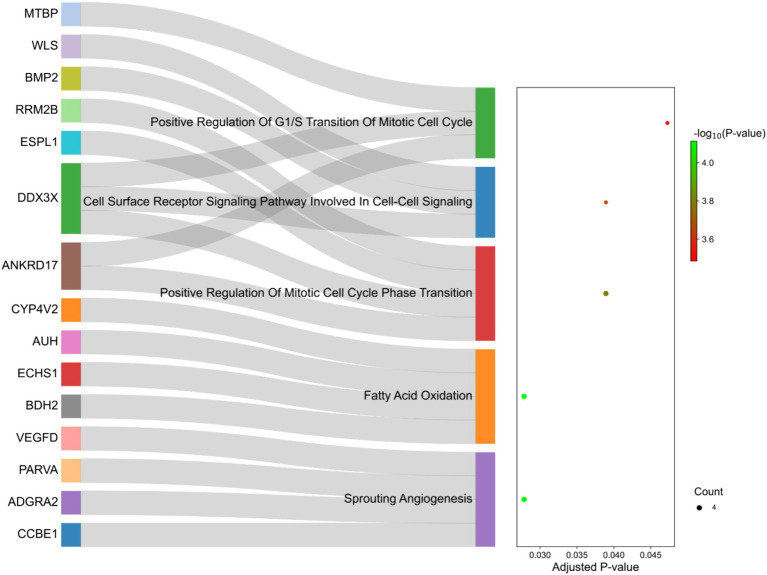
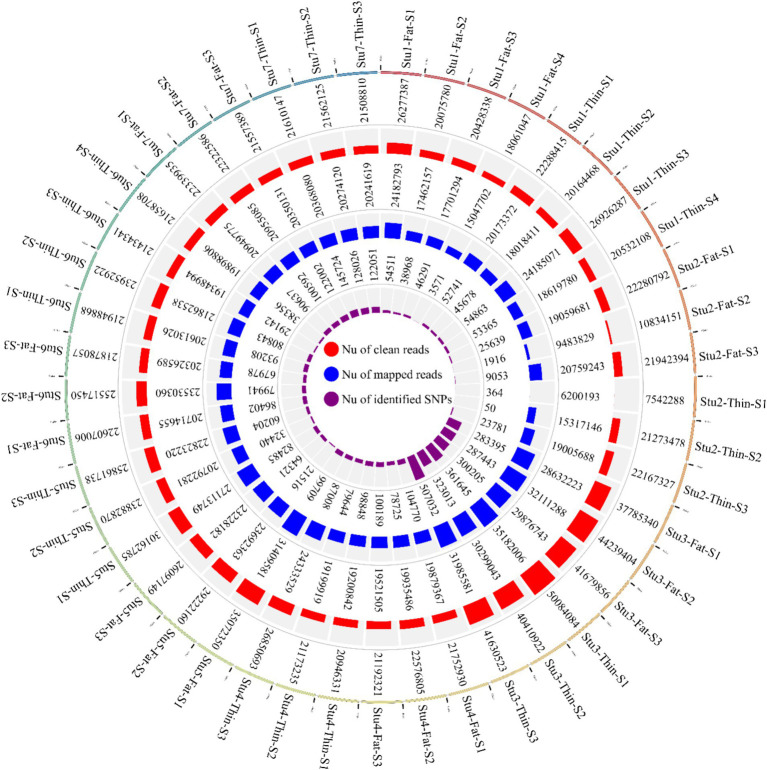
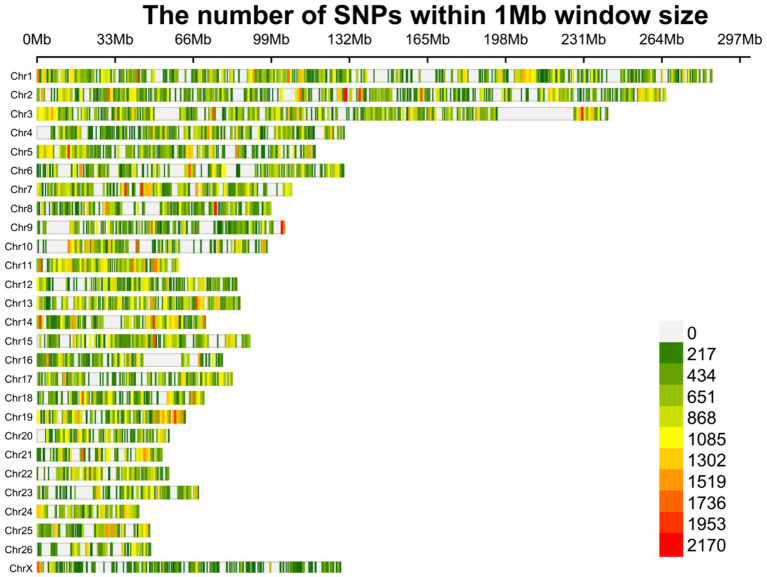
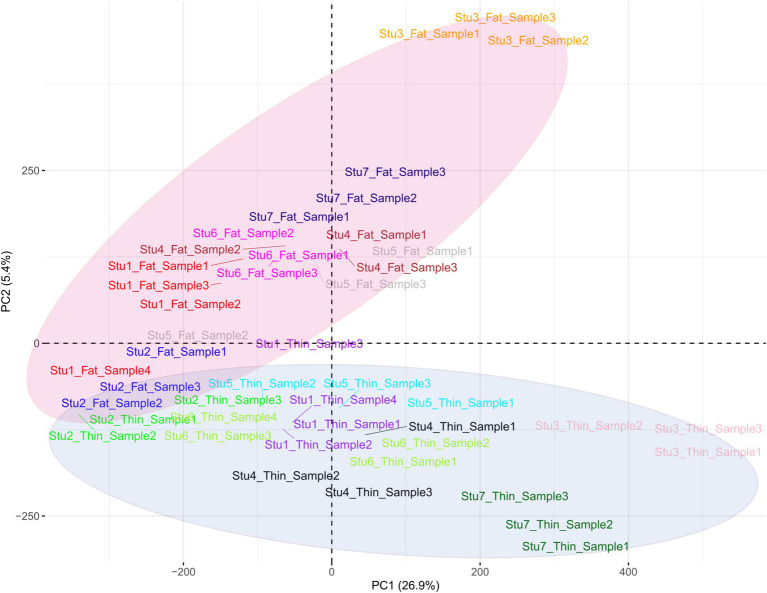
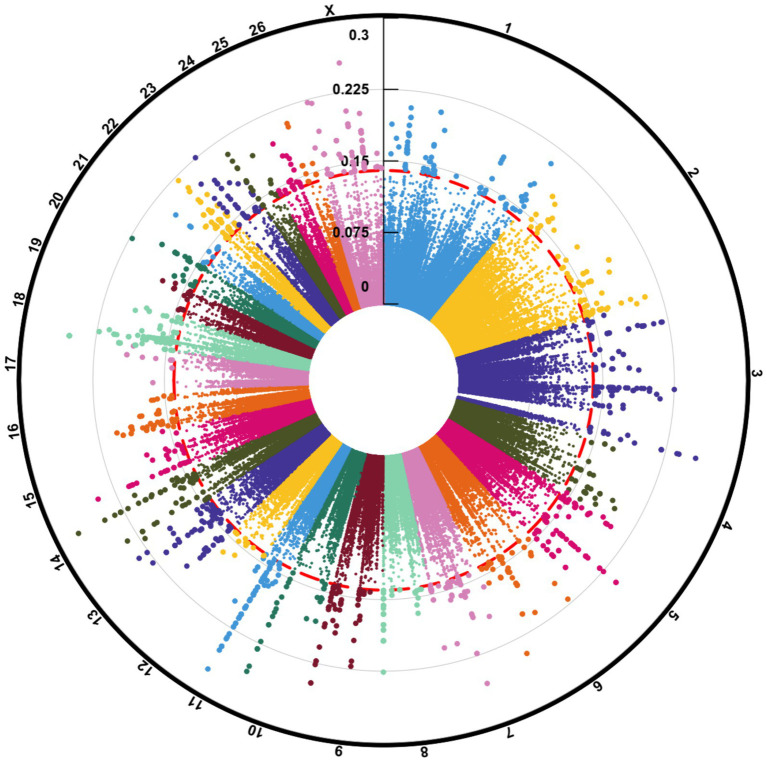
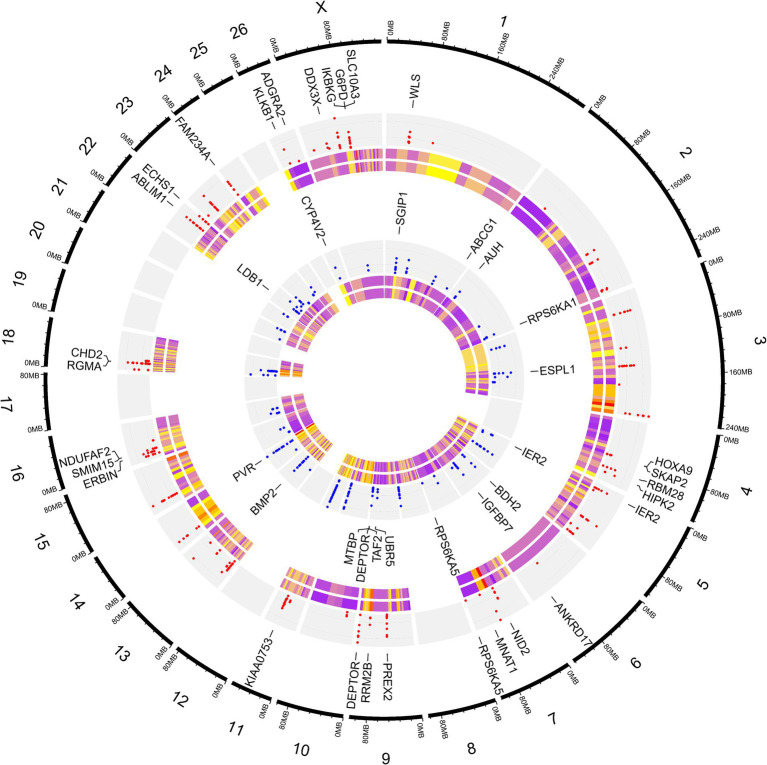
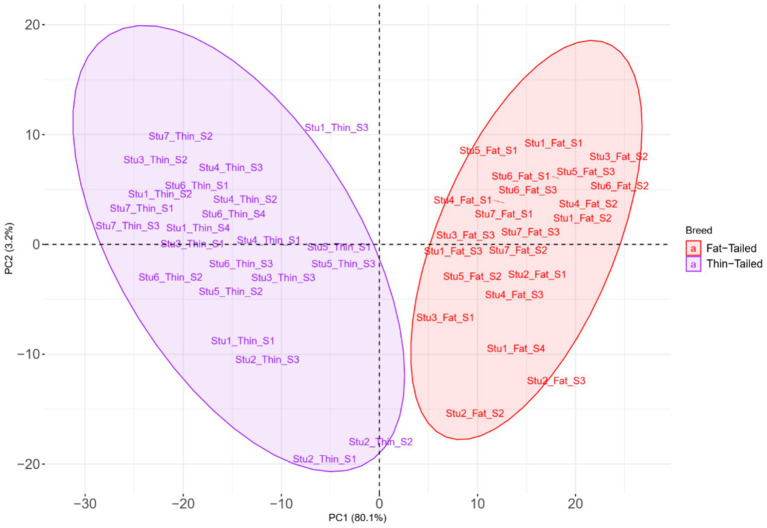
Understanding the genetic background behind fat-tail development in sheep can be useful to develop breeding programs for genetic improvement, while the genetic basis of fat-tail formation is still not well understood. Here, to identify genomic regions influencing fat-tail size in sheep, a comprehensive selection signature identification analysis was performed through comparison of fat- and thin-tailed sheep breeds. Furthermore, to gain the first insights into the potential use of RNA-Seq for selection signature identification analysis, SNP calling was performed using RNA-Seq datasets. In total, 45 RNA-Seq samples from seven cohort studies were analyzed, and the F method was used to detect selection signatures. Our findings indicated that RNA-Seq could be of potential utility for selection signature identification analysis. In total, 877 SNPs related to 103 genes were found to be under selection in 92 genomic regions. Functional annotation analysis reinforced the hypothesis that genes involved in fatty acid oxidation May modulate fat accumulation in the tail of sheep and highlighted the potential regulatory role of angiogenesis process in the fat deposition. In agreement with most previous studies, our results re-emphasize that the gene is targeted by selection during sheep evolution. Further gene annotation analysis of the regions targeted by the sheep evolution process revealed that a large number of genes included in these regions are directly associated with fat metabolism, including those previously reported as candidates involved in sheep fat-tail morphology, such as , , and . Moreover, a number of genes, including , , , and were of particular interest because they are well-known fat metabolism-associated genes and are considered novel candidates involved in fat-tail size. Consistent with the selection signature identification analysis, principal component analysis clustered the samples into two completely separate groups according to fat- and thin-tailed breeds. Our results provide novel insights into the genomic basis of phenotypic diversity related to the fat-tail of sheep breeds and can be used to determine directions for improving breeding strategies in the future.
了解绵羊肥尾发育背后的遗传背景,对于制定遗传改良育种计划可能会有所帮助,然而肥尾形成的遗传基础仍未得到充分理解。在此,为了鉴定影响绵羊肥尾大小的基因组区域,通过比较肥尾和瘦尾绵羊品种进行了全面的选择信号鉴定分析。此外,为了初步了解RNA测序在选择信号鉴定分析中的潜在用途,使用RNA测序数据集进行了单核苷酸多态性(SNP)检测。总共分析了来自七项队列研究的45个RNA测序样本,并使用F方法检测选择信号。我们的研究结果表明,RNA测序在选择信号鉴定分析中可能具有潜在用途。总共在92个基因组区域中发现了与103个基因相关的877个SNP处于选择状态。功能注释分析强化了这样一种假设,即参与脂肪酸氧化的基因可能调节绵羊尾部的脂肪积累,并突出了血管生成过程在脂肪沉积中的潜在调节作用。与大多数先前的研究一致,我们的结果再次强调该基因在绵羊进化过程中受到选择。对绵羊进化过程所靶向区域的进一步基因注释分析表明,这些区域中包含的大量基因与脂肪代谢直接相关,包括那些先前被报道为参与绵羊肥尾形态的候选基因,如 、 、 和 。此外,一些基因,包括 、 、 、 和 特别受关注,因为它们是众所周知的与脂肪代谢相关的基因,并且被认为是参与肥尾大小的新候选基因。与选择信号鉴定分析一致,主成分分析根据肥尾和瘦尾品种将样本聚为两个完全分开的组。我们的结果为绵羊品种肥尾相关表型多样性的基因组基础提供了新的见解,并可用于确定未来改进育种策略的方向。