Earlham Institute, Norwich Research Park, Norwich, NR4 7UZ, UK.
Department of Metabolism, Digestion and Reproduction, Imperial College London, The Commonwealth Building, The Hammersmith Hospital, Du Cane Road, London, W12 0NN, UK.
BMC Bioinformatics. 2024 Oct 18;25(1):334. doi: 10.1186/s12859-024-05948-7.
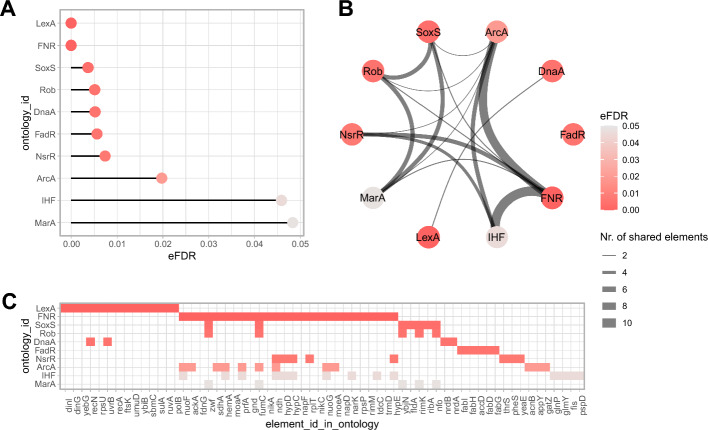
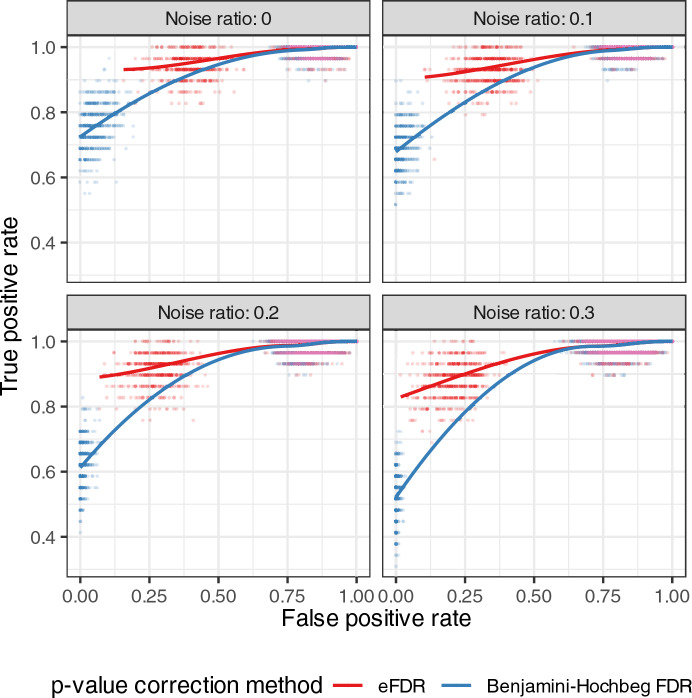
Traditional gene set enrichment analyses are typically limited to a few ontologies and do not account for the interdependence of gene sets or terms, resulting in overcorrected p-values. To address these challenges, we introduce mulea, an R package offering comprehensive overrepresentation and functional enrichment analysis. mulea employs a progressive empirical false discovery rate (eFDR) method, specifically designed for interconnected biological data, to accurately identify significant terms within diverse ontologies. mulea expands beyond traditional tools by incorporating a wide range of ontologies, encompassing Gene Ontology, pathways, regulatory elements, genomic locations, and protein domains. This flexibility enables researchers to tailor enrichment analysis to their specific questions, such as identifying enriched transcriptional regulators in gene expression data or overrepresented protein domains in protein sets. To facilitate seamless analysis, mulea provides gene sets (in standardised GMT format) for 27 model organisms, covering 22 ontology types from 16 databases and various identifiers resulting in almost 900 files. Additionally, the muleaData ExperimentData Bioconductor package simplifies access to these pre-defined ontologies. Finally, mulea's architecture allows for easy integration of user-defined ontologies, or GMT files from external sources (e.g., MSigDB or Enrichr), expanding its applicability across diverse research areas. mulea is distributed as a CRAN R package downloadable from https://cran.r-project.org/web/packages/mulea/ and https://github.com/ELTEbioinformatics/mulea . It offers researchers a powerful and flexible toolkit for functional enrichment analysis, addressing limitations of traditional tools with its progressive eFDR and by supporting a variety of ontologies. Overall, mulea fosters the exploration of diverse biological questions across various model organisms.
传统的基因集富集分析通常仅限于少数本体论,并且不考虑基因集或术语的相互依存关系,导致校正过度的 p 值。为了解决这些挑战,我们引入了 mulea,这是一个提供全面过表达和功能富集分析的 R 包。mulea 采用了一种渐进式经验假发现率(eFDR)方法,专门为相互关联的生物数据设计,以准确识别不同本体论中的显著术语。mulea 通过纳入广泛的本体论,超越了传统工具,包括基因本体论、途径、调节元件、基因组位置和蛋白质结构域。这种灵活性使研究人员能够根据自己的具体问题定制富集分析,例如识别基因表达数据中丰富的转录调节剂或蛋白质集中过表达的蛋白质结构域。为了方便无缝分析,mulea 提供了 27 种模式生物的基因集(以标准化的 GMT 格式),涵盖了 16 个数据库中的 22 种本体论类型和各种标识符,总共产生了近 900 个文件。此外,muleaData ExperimentData Bioconductor 包简化了对这些预定义本体论的访问。最后,mulea 的架构允许轻松集成用户定义的本体论或来自外部源(例如,MSigDB 或 Enrichr)的 GMT 文件,从而扩展了其在各个研究领域的适用性。mulea 作为一个 CRAN R 包发布,可以从 https://cran.r-project.org/web/packages/mulea/ 和 https://github.com/ELTEbioinformatics/mulea 下载。它为研究人员提供了一个功能强大且灵活的功能富集分析工具包,通过渐进式 eFDR 解决了传统工具的局限性,并支持各种本体论。总体而言,mulea 促进了对各种模型生物中不同生物学问题的探索。