Sołtyka-Krajewska Maja, Ziemniak Marcin, Zawadzka-Kazimierczuk Anna, Skrzypczyk Paulina, Siwiak-Niedbalska Ewelina, Jaśkiewicz Anna, Zieliński Rafał, Fokt Izabela, Skóra Stanisław, Koźmiński Wiktor, Woźniak Krzysztof, Priebe Waldemar, Pająk-Tarnacka Beata
Department of Medical Biology, Kaczkowski Military Institute of Hygiene and Epidemiology, Kozielska 4, 01-163 Warsaw, Poland.
Biological and Chemical Research Centre, Department of Chemistry, University of Warsaw, Żwirki i Wigury 101, 02-089 Warsaw, Poland.
Biomedicines. 2024 Oct 1;12(10):2240. doi: 10.3390/biomedicines12102240.
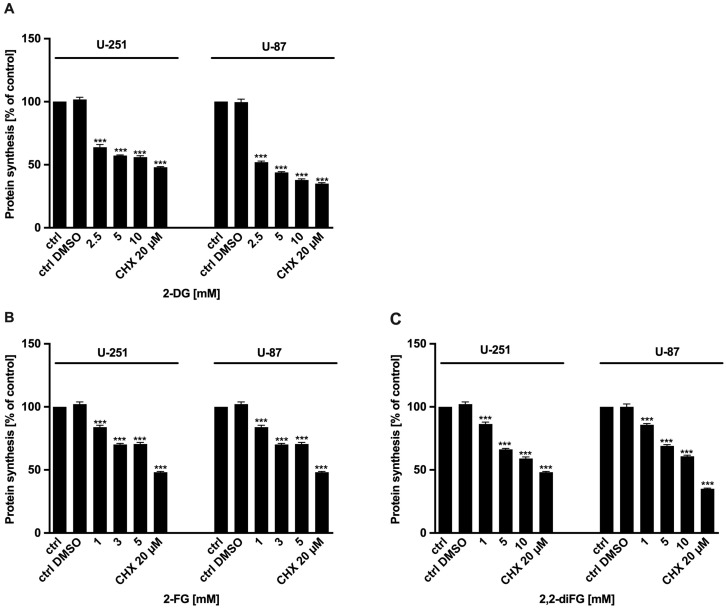
One defining feature of various aggressive cancers, including glioblastoma multiforme (GBM), is glycolysis upregulation, making its inhibition a promising therapeutic approach. One promising compound is 2-deoxy-d-glucose (2-DG), a d-glucose analog with high clinical potential due to its ability to inhibit glycolysis. Upon uptake, 2-DG is phosphorylated by hexokinase to 2-DG-6-phosphate, which inhibits hexokinase and downstream glycolytic enzymes. Unfortunately, therapeutic use of 2-DG is limited by poor pharmacokinetics, suppressing its efficacy.
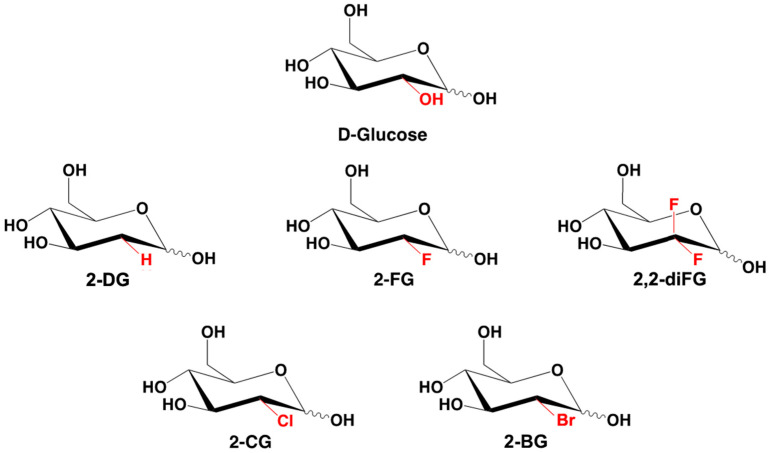
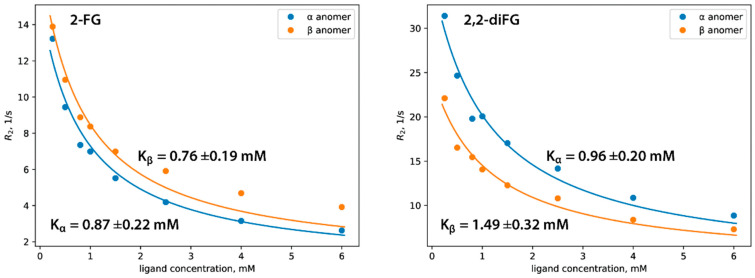
To address these issues, we synthesized novel halogenated 2-DG analogs (2-FG, 2,2-diFG, 2-CG, and 2-BG) and evaluated their glycolytic inhibition in GBM cells. Our in vitro and computational studies suggest that these derivatives modulate hexokinase activity differently.
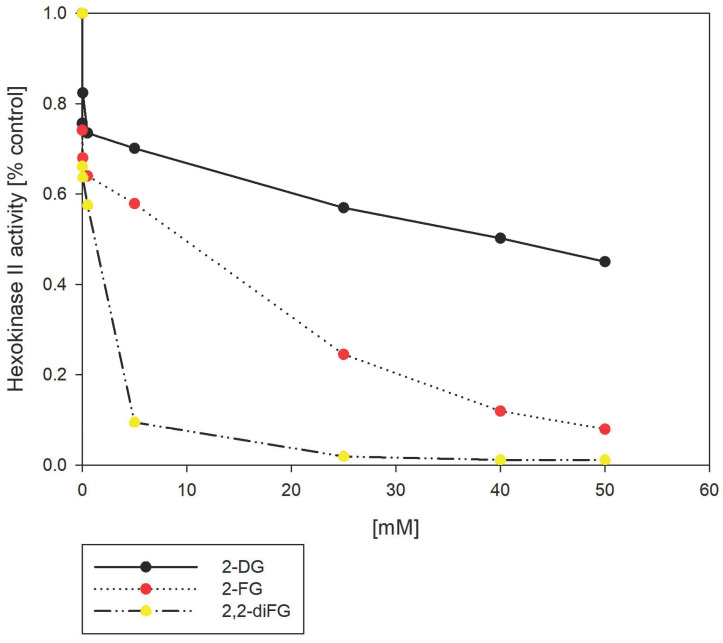
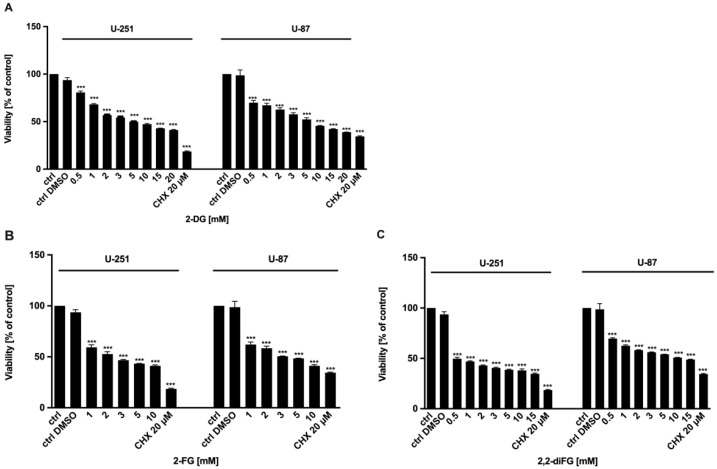
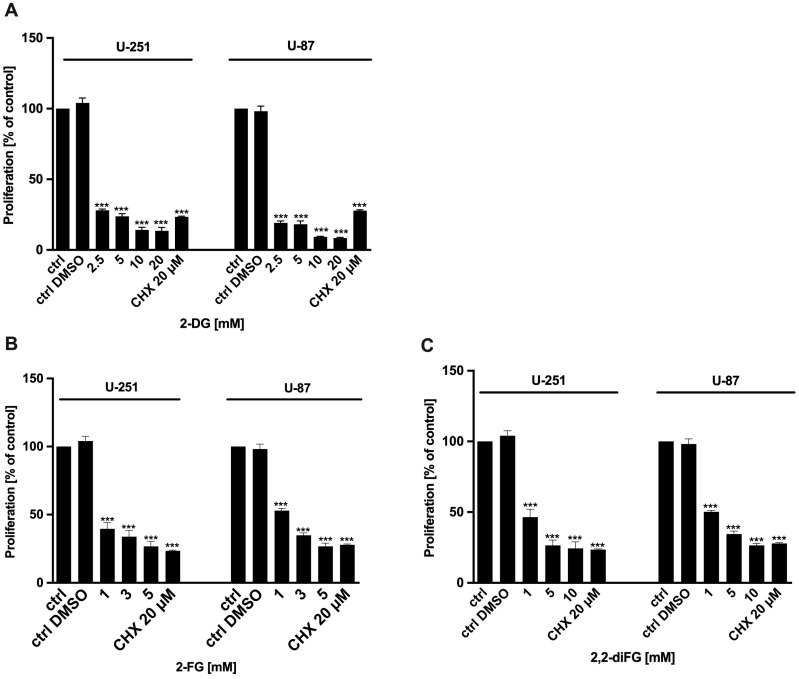
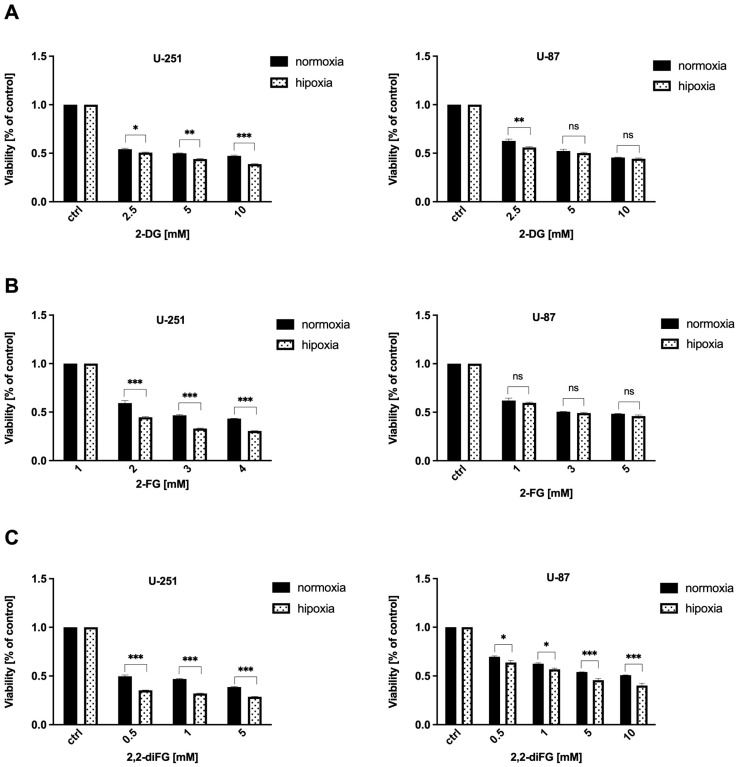
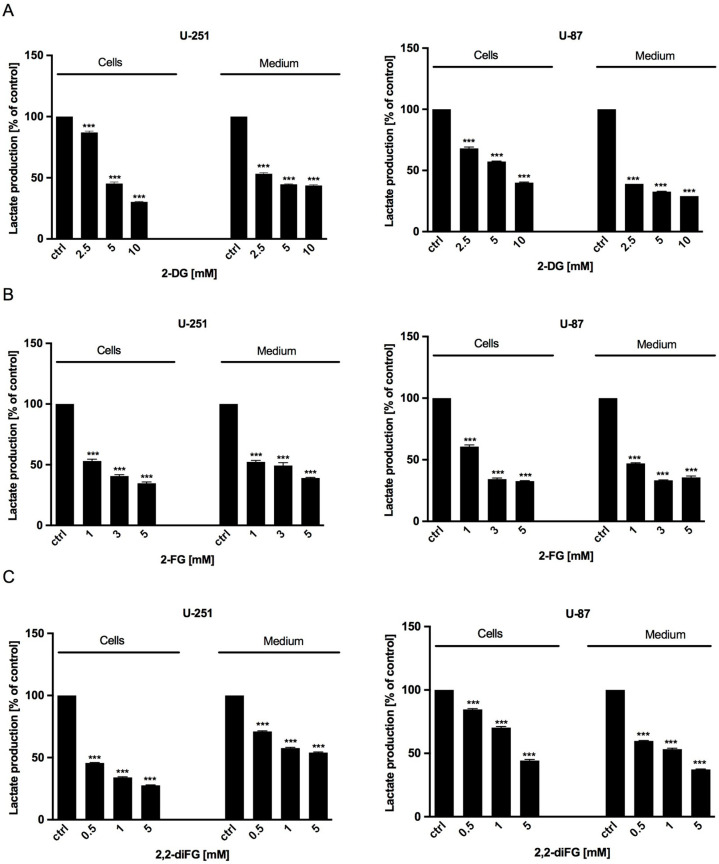
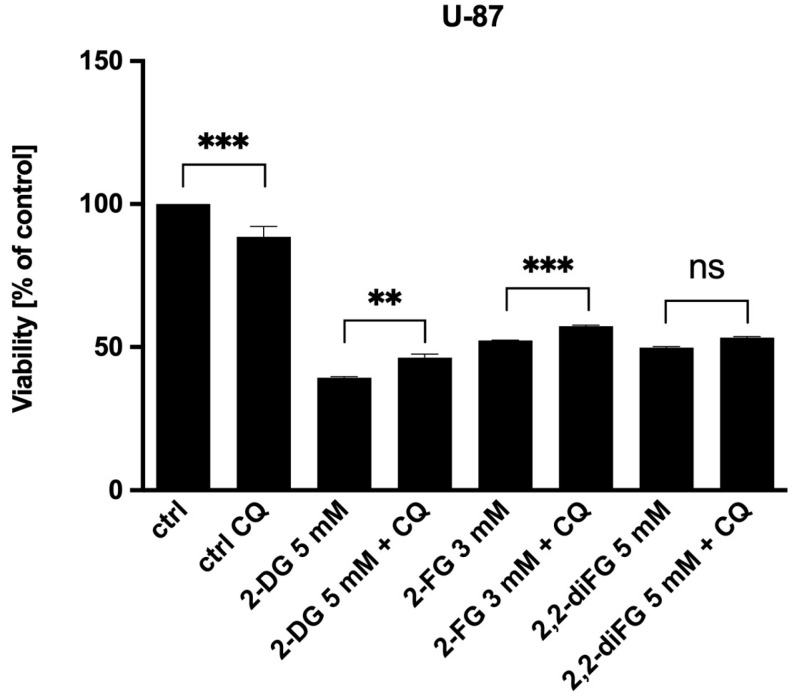
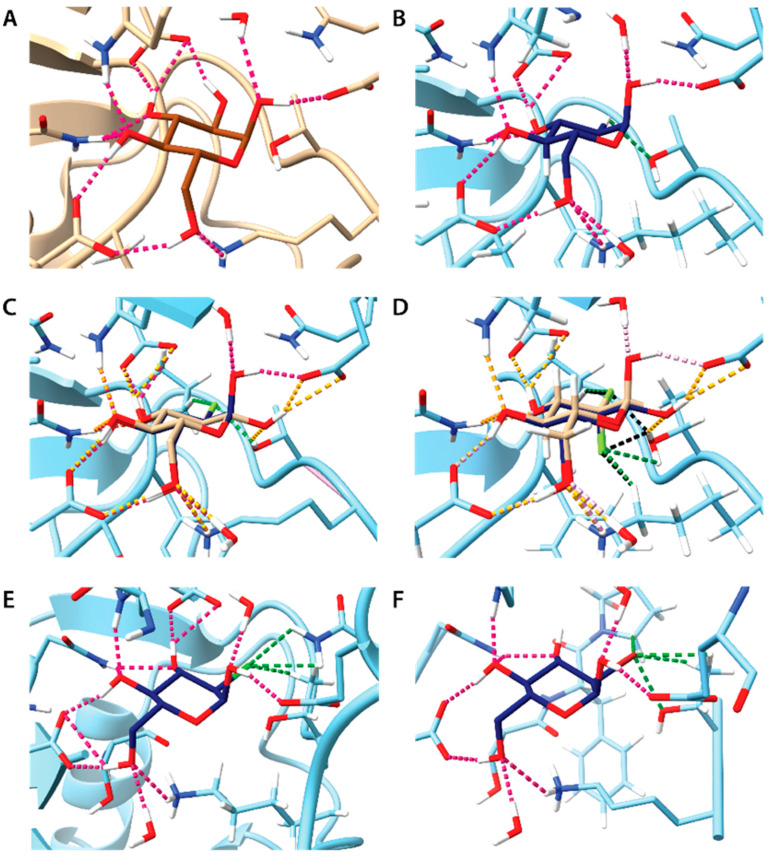
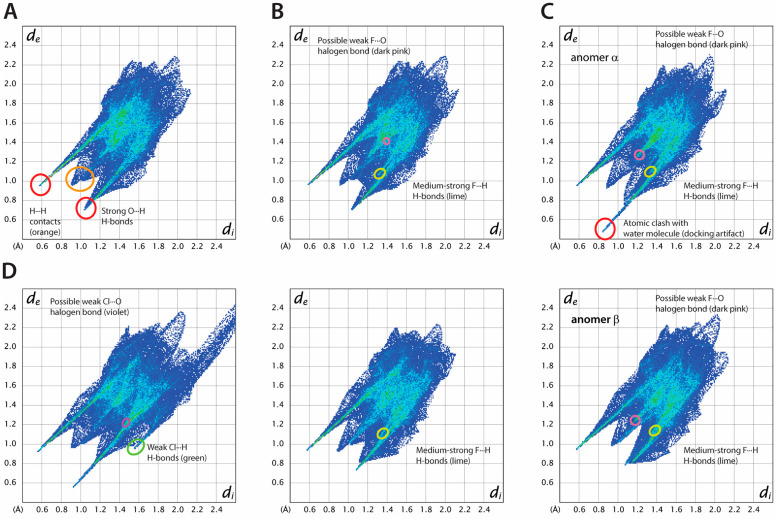
Fluorinated compounds show the most potent cytotoxic effects, indicated by the lowest IC values. These effects were more pronounced in hypoxic conditions. F NMR experiments and molecular docking confirmed that fluorinated derivatives bind hexokinase comparably to glucose. Enzymatic assays demonstrated that all halogenated derivatives are more effective HKII inhibitors than 2-DG, particularly through their 6-phosphates. By modifying the C-2 position with halogens, these compounds may overcome the poor pharmacokinetics of 2-DG. The modifications seem to enhance the stability and uptake of the compounds, making them effective at lower doses and over prolonged periods.
This research has the potential to reshape the treatment landscape for GBM and possibly other cancers by offering a more targeted, effective, and metabolically focused therapeutic approach. The application of halogenated 2-DG analogs represents a promising advancement in cancer metabolism-targeted therapies, with the potential to overcome current treatment limitations.
包括多形性胶质母细胞瘤(GBM)在内的各种侵袭性癌症的一个显著特征是糖酵解上调,因此抑制糖酵解是一种很有前景的治疗方法。一种有前景的化合物是2-脱氧-D-葡萄糖(2-DG),它是一种D-葡萄糖类似物,因其抑制糖酵解的能力而具有很高的临床潜力。摄入后,2-DG被己糖激酶磷酸化为2-DG-6-磷酸,后者抑制己糖激酶和下游糖酵解酶。不幸的是,2-DG的治疗用途受到不良药代动力学的限制,从而抑制了其疗效。
为了解决这些问题,我们合成了新型卤代2-DG类似物(2-FG、2,2-二FG、2-CG和2-BG),并评估了它们对GBM细胞糖酵解的抑制作用。我们的体外和计算研究表明,这些衍生物对己糖激酶活性的调节方式不同。
氟化化合物显示出最有效的细胞毒性作用,以最低的IC值为指标。这些作用在缺氧条件下更为明显。19F NMR实验和分子对接证实,氟化衍生物与己糖激酶的结合与葡萄糖相当。酶活性测定表明,所有卤代衍生物都是比2-DG更有效的HKII抑制剂,特别是通过它们的6-磷酸酯。通过用卤素修饰C-2位置,这些化合物可能克服2-DG不良的药代动力学。这些修饰似乎增强了化合物的稳定性和摄取,使其在较低剂量和较长时间内有效。
这项研究有可能通过提供一种更有针对性、更有效且以代谢为重点的治疗方法,重塑GBM以及可能其他癌症的治疗格局。卤代2-DG类似物的应用代表了癌症代谢靶向治疗方面有前景的进展,有可能克服当前的治疗局限性。