Lishui Key Laboratory of Brain Health and Severe Brain Disorders, Department of Rehabilitation & Clinical Laboratory, Lishui Second People's Hospital, Lishui, China.
Key Laboratory of Cell Engineering in Guizhou Province, Affiliated Hospital of Zunyi Medical University, Zunyi, China.
BMC Neurol. 2024 Oct 31;24(1):423. doi: 10.1186/s12883-024-03926-3.
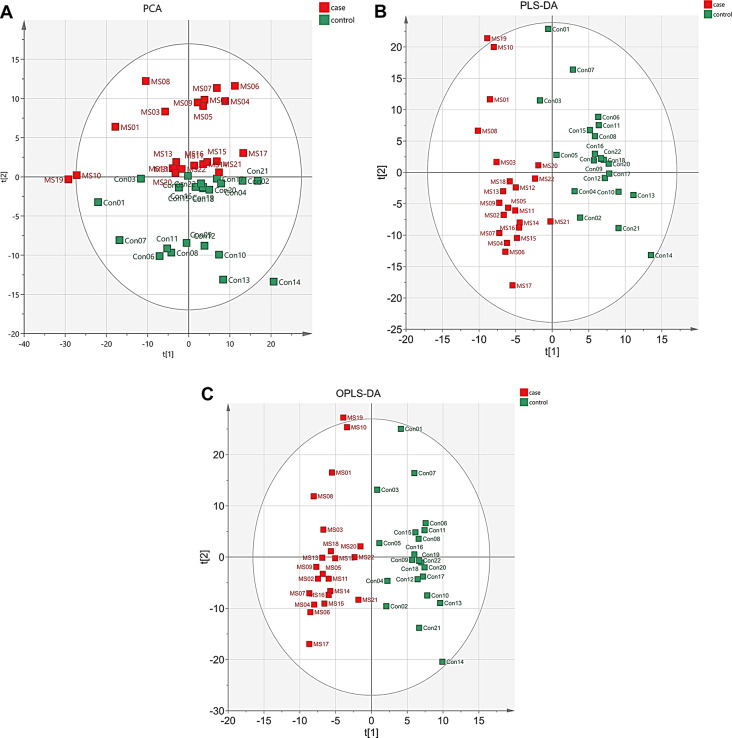
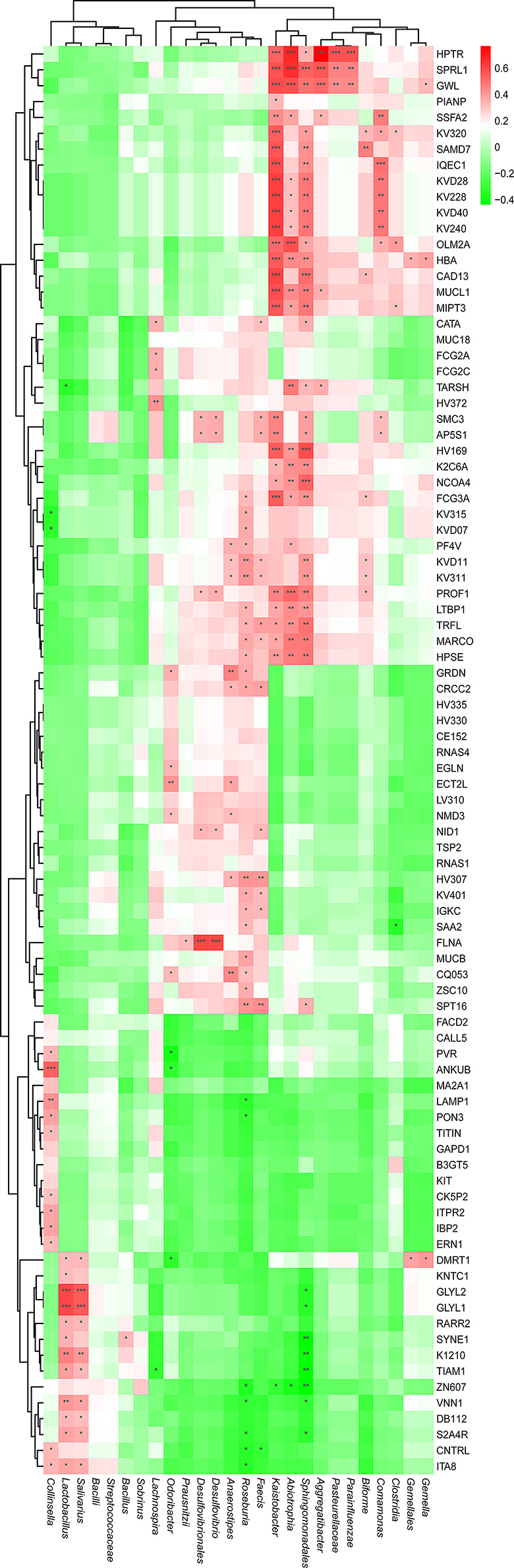
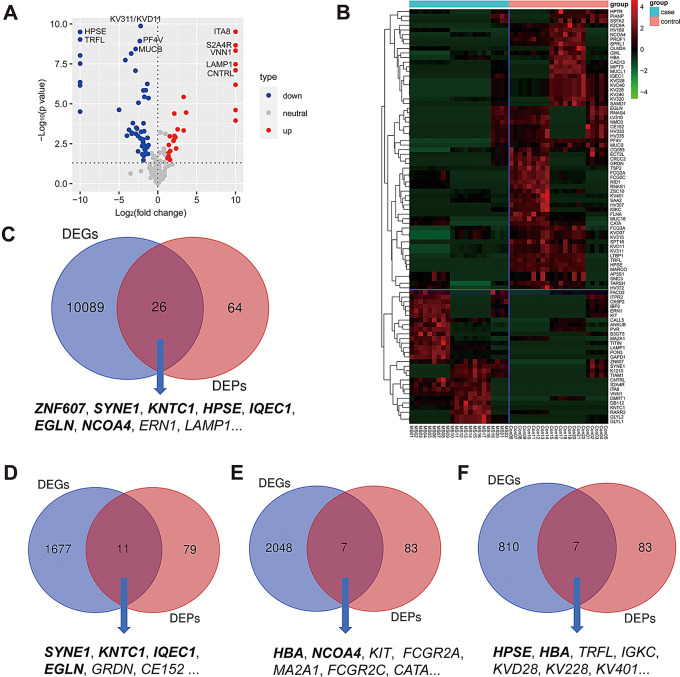
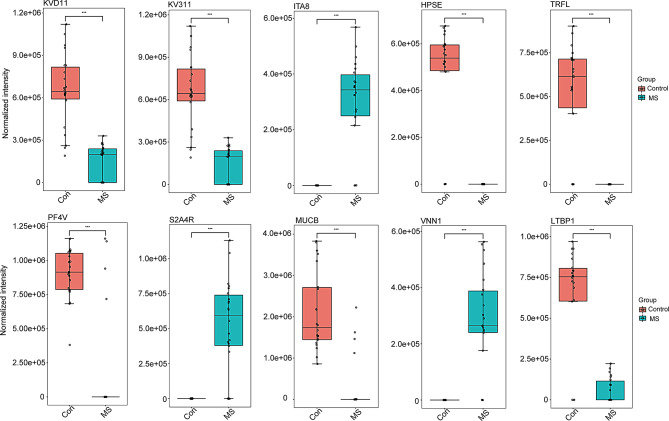
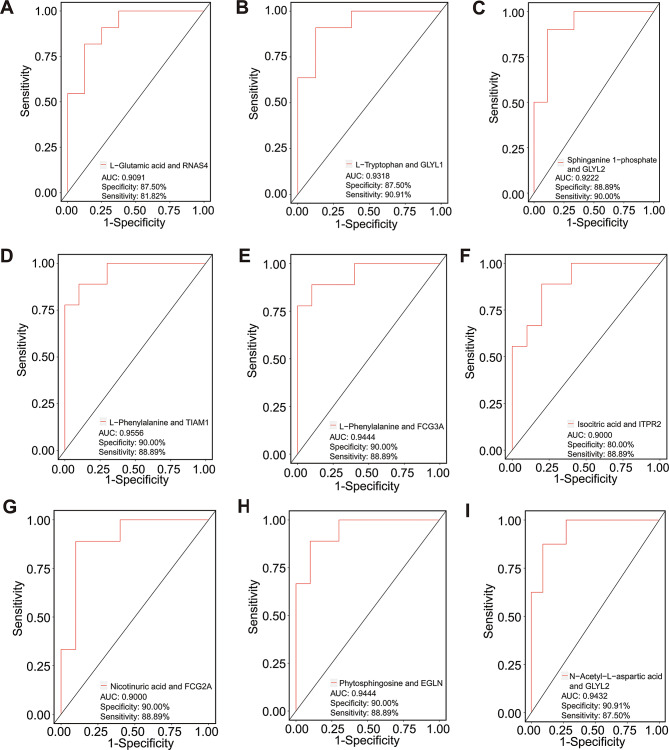
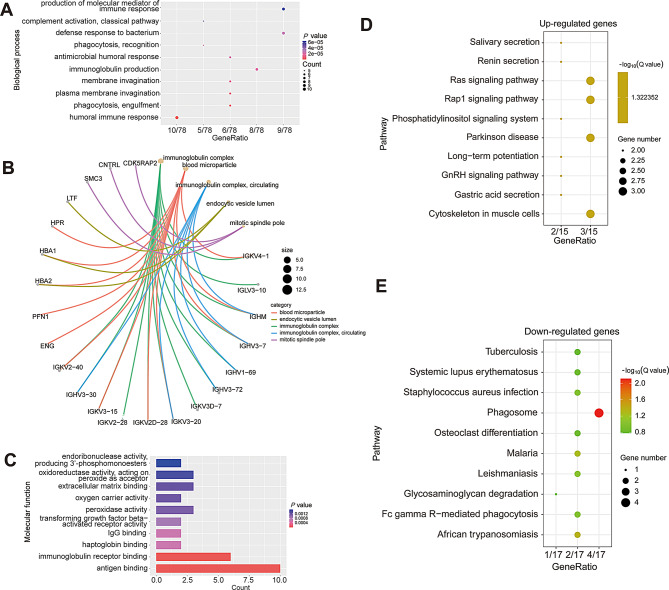
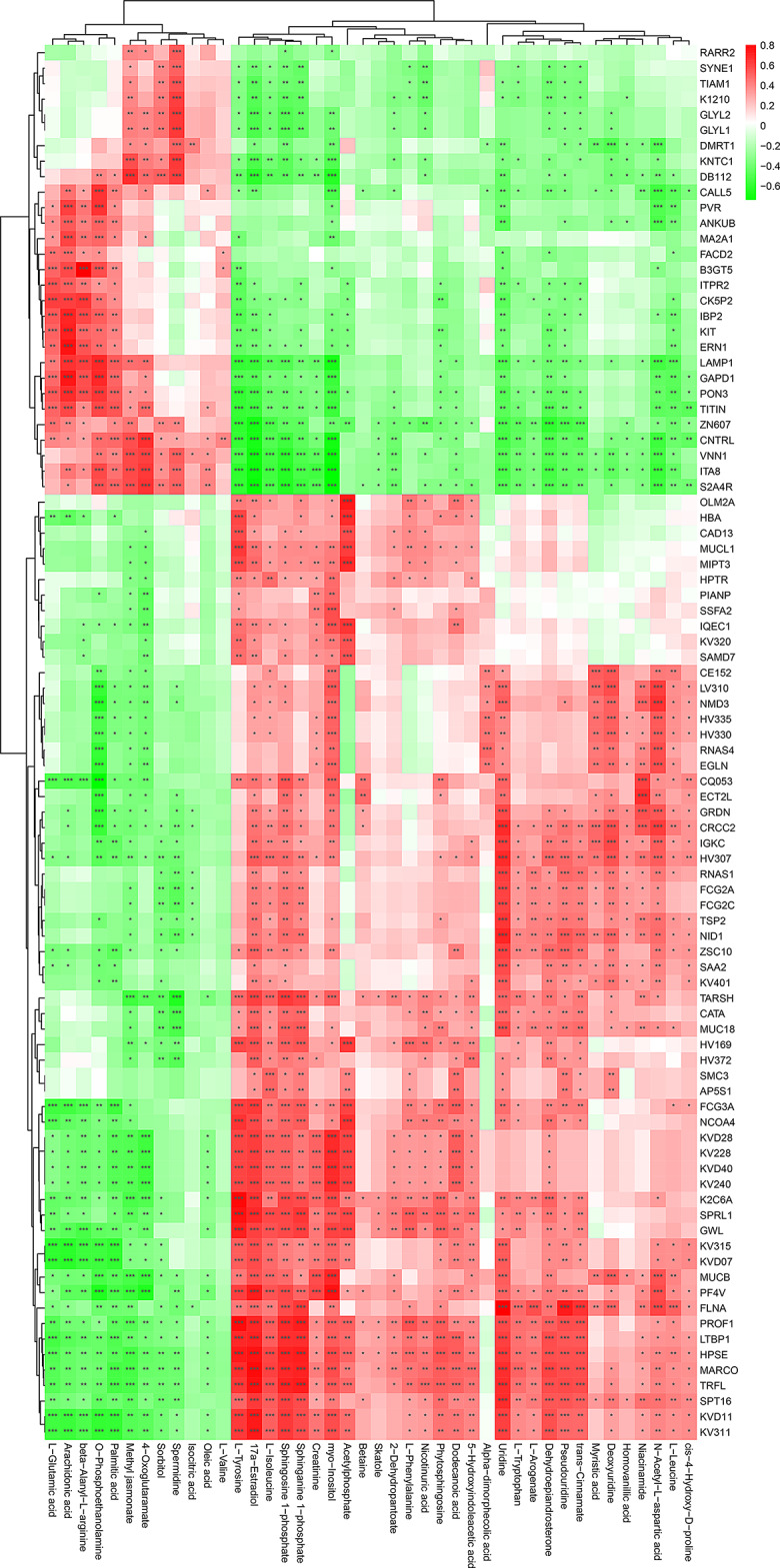
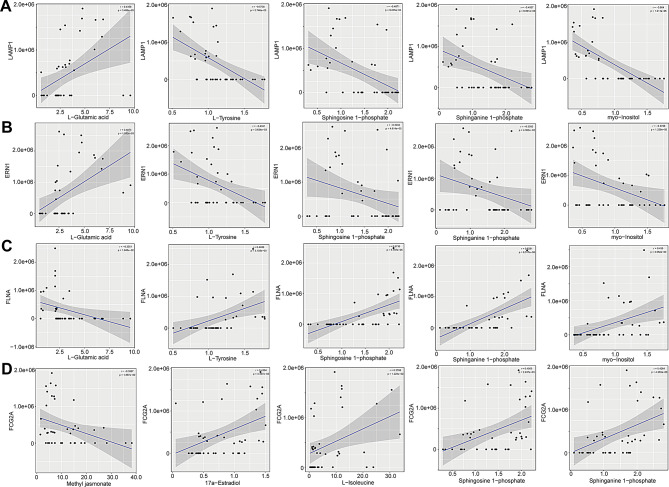
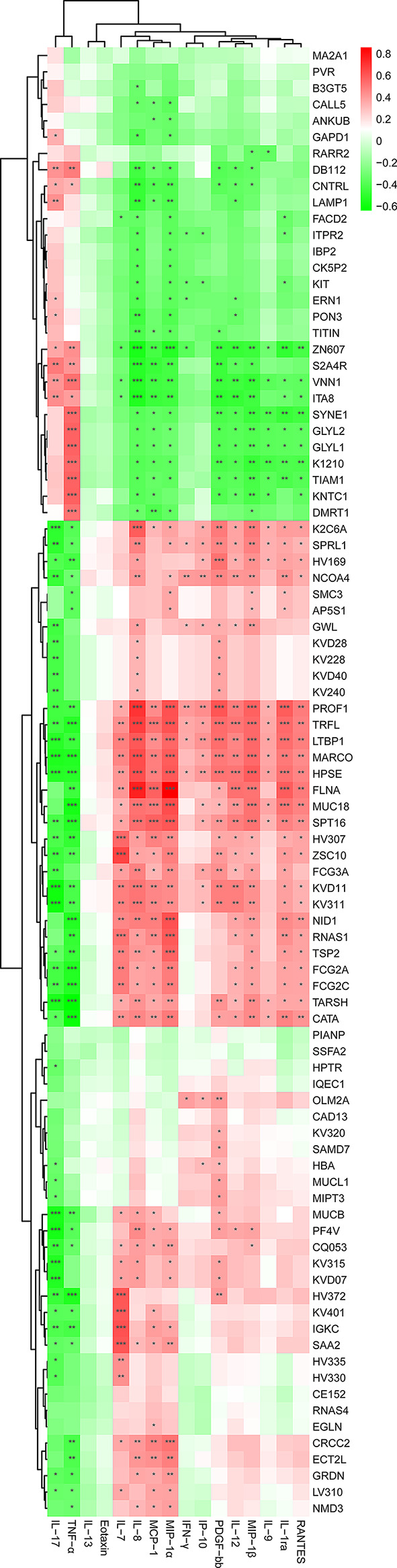
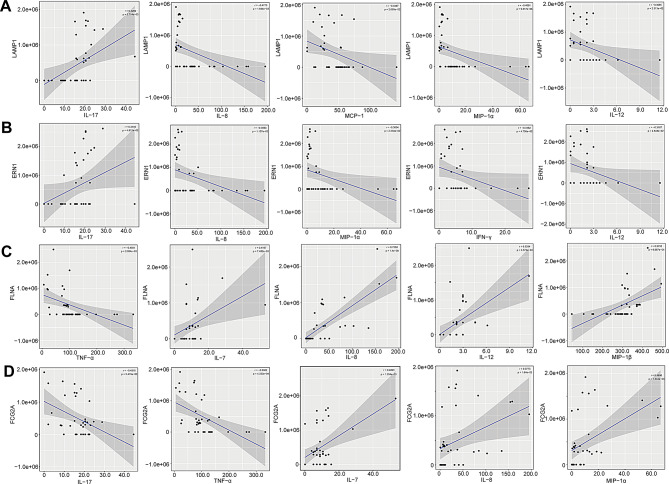
Multiple sclerosis (MS) is an autoimmune disorder caused by chronic inflammatory reactions in the central nervous system. Currently, little is known about the changes of plasma proteomic profiles in Chinese patients with MS (CpwMS) and its relationship with the altered profiles of multi-omics such as metabolomics and gut microbiome, as well as potential molecular networks that underlie the etiology of MS. To uncover the characteristics of proteomics landscape and potential multi-omics interaction networks in CpwMS, Plasma samples were collected from 22 CpwMS and 22 healthy controls (HCs) and analyzed using a Tandem Mass Tag (TMT)-based quantitative proteomics approach. Our results showed that the plasma proteomics pattern was significantly different in CpwMS compared to HCs. A total of 90 differentially expressed proteins (DEPs), such as LAMP1 and FCG2A, were identified in CpwMS plasma comparing to HCs. Furthermore, we also observed extensive and significant correlations between the altered proteomic profiles and the changes of metabolome, gut microbiome, as well as altered immunoinflammatory responses in MS-affected patients. For instance, the level of LAMP1 and ERN1 were significantly and positively correlated with the concentrations of metabolite L-glutamic acid and pro-inflammatory factor IL-17 (Padj < 0.05). However, they were negatively correlated with the amounts of other metabolites such as L-tyrosine and sphingosine 1-phosphate, as well as the concentrations of IL-8 and MIP-1α. This study outlined the underlying multi-omics integrated mechanisms that might regulate peripheral immunoinflammatory responses and MS progression. These findings are potentially helpful for developing new assisting diagnostic biomarker and therapeutic strategies for MS.
多发性硬化症(MS)是一种由中枢神经系统慢性炎症反应引起的自身免疫性疾病。目前,人们对中国多发性硬化症患者(CpwMS)的血浆蛋白质组谱变化及其与代谢组学和肠道微生物组等多组学改变以及潜在分子网络的关系知之甚少,这些潜在分子网络是 MS 发病机制的基础。为了揭示 CpwMS 中蛋白质组学特征和潜在的多组学相互作用网络,我们从 22 名 CpwMS 和 22 名健康对照(HC)中收集血浆样本,并使用串联质量标签(TMT)定量蛋白质组学方法进行分析。我们的研究结果表明,与 HCs 相比,CpwMS 中的血浆蛋白质组学模式明显不同。与 HCs 相比,CpwMS 血浆中鉴定出 90 个差异表达蛋白(DEPs),如 LAMP1 和 FCG2A。此外,我们还观察到改变的蛋白质组学图谱与代谢组、肠道微生物组以及 MS 患者免疫炎症反应改变之间存在广泛而显著的相关性。例如,LAMP1 和 ERN1 的水平与代谢物 L-谷氨酸和促炎因子 IL-17 的浓度呈显著正相关(Padj<0.05)。然而,它们与其他代谢物如 L-酪氨酸和鞘氨醇 1-磷酸的含量以及 IL-8 和 MIP-1α 的浓度呈负相关。这项研究概述了可能调节外周免疫炎症反应和 MS 进展的潜在多组学综合机制。这些发现可能有助于开发用于 MS 的新辅助诊断生物标志物和治疗策略。