Department of Pharmacy, the Second Xiangya Hospital, Central South University, Changsha, 410011, China.
Department of Pharmacy, Wuzhou Gongren Hospital, Wuzhou, 543000, China.
Cell Biol Toxicol. 2024 Nov 6;40(1):94. doi: 10.1007/s10565-024-09930-0.
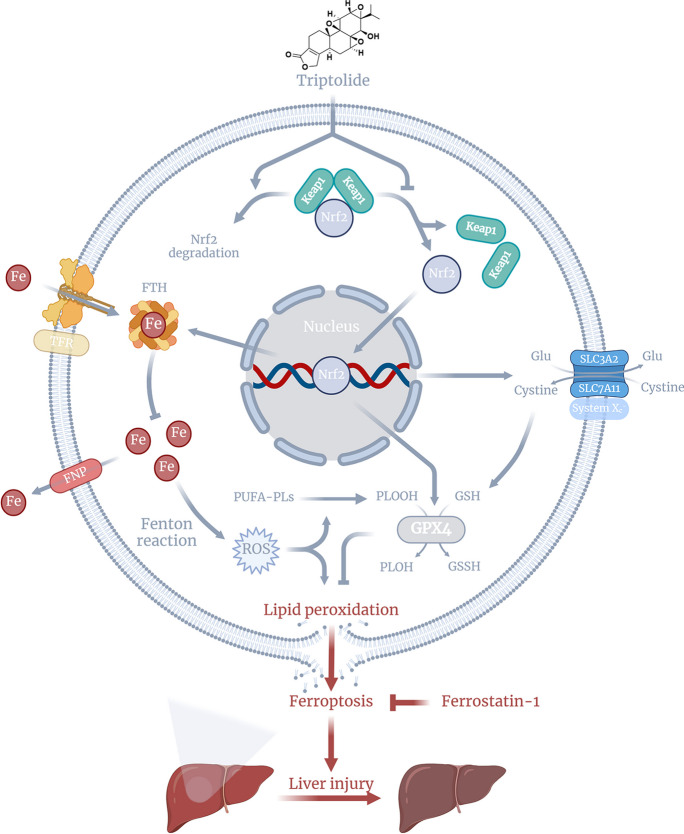
Triptolide (TP), a principal active substance from Tripterygium wilfordii, exhibits various pharmacological effects. However, its potential hepatotoxicity has always been a significant concern in clinical applications.
This research aimed to explore the involvement of ferroptosis in TP-mediated hepatic injury and the underlying mechanisms.
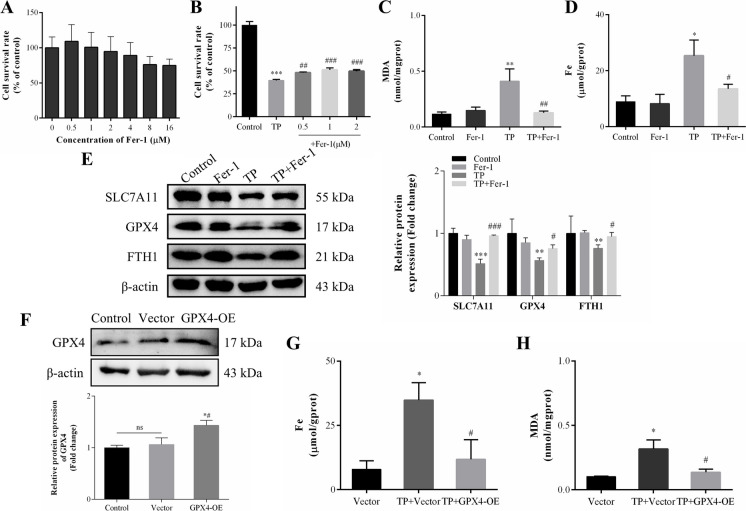
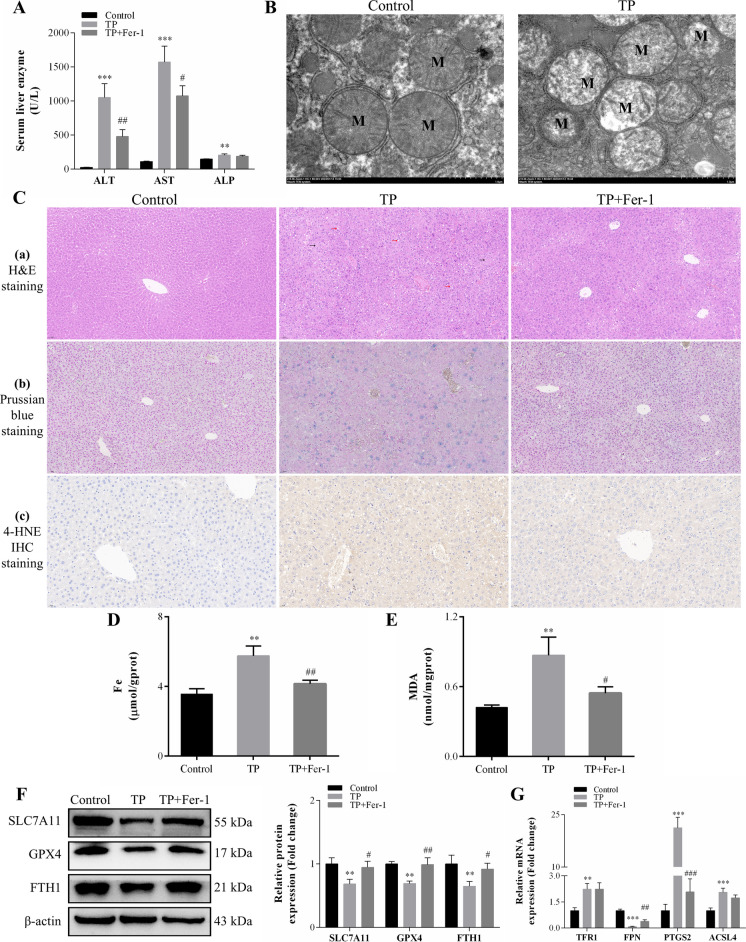
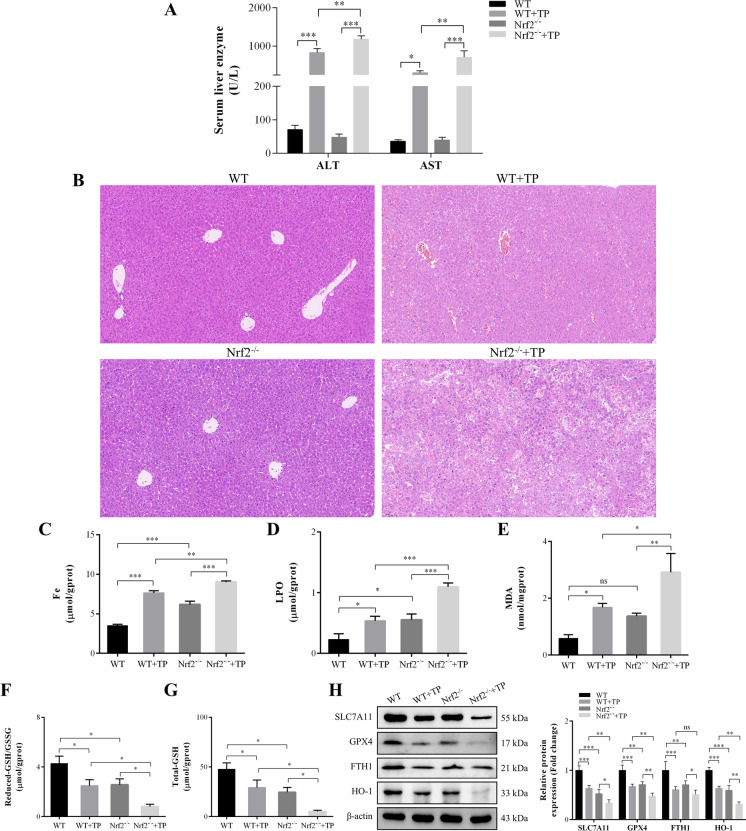
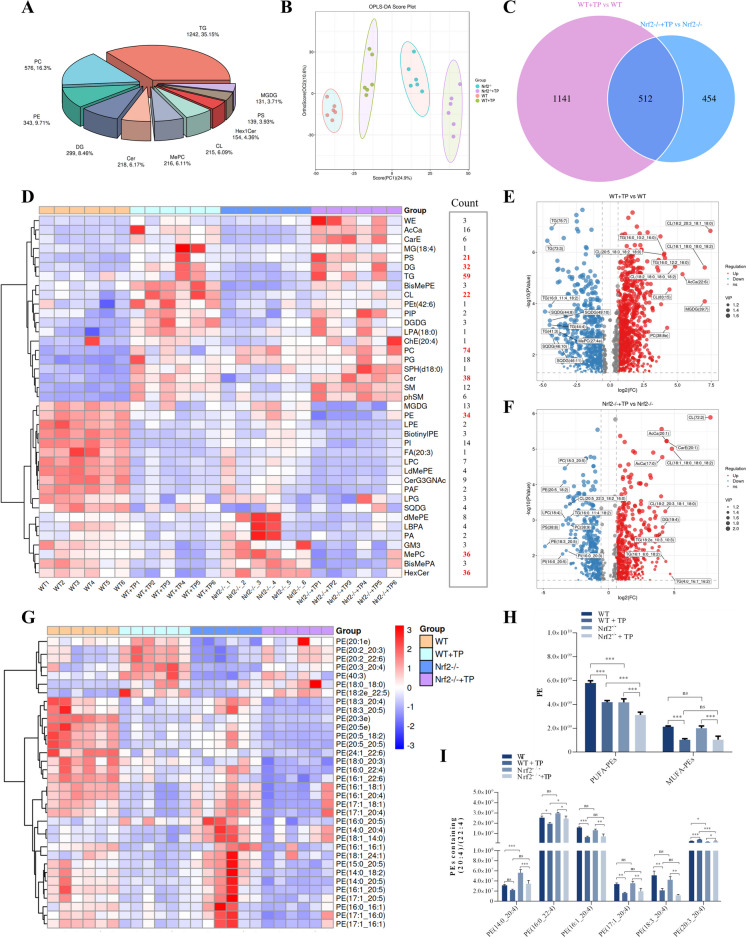
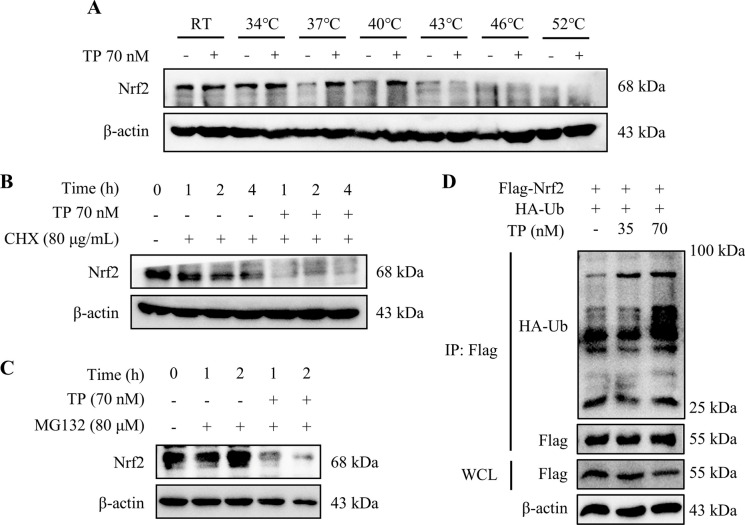
In this study, in vitro and in vivo experiments were involved. Hepatocyte damage caused by TP was evaluated using MTT assays, liver enzyme measurement and H&E staining technique. Ferroptosis was assessed by measuring iron level, lipid peroxide, glutathione (GSH), mitochondrial morphology and the key protein/mRNA expression implicated in ferroptosis. To verify the contribution of ferroptosis to TP-induced liver damage, the ferroptosis inhibitor Ferrostatin-1 (Fer-1) and a plasmid for overexpressing glutathione peroxidase 4 (GPX4) were employed. Subsequently, nuclear factor erythroid 2-related factor 2 (Nrf2) knockout mice and Nrf2 overexpression plasmid were utilized to investigate the underlying mechanisms. Nontargeted lipidomics was used to analyze lipid metabolism in mouse liver. Moreover, the cellular thermal shift assay (CETSA), cycloheximide (CHX) and MG132 treatments, and immunoprecipitation (IP) assays were applied to validate the binding of TP to Nrf2 and their interactions.
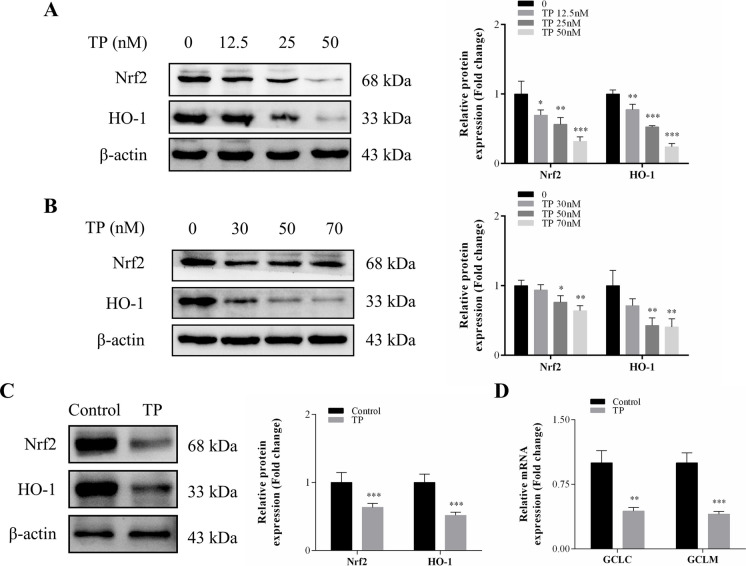
TP triggered ferroptosis in hepatocytes, as indicated by iron accumulation and lipid peroxidation. Ferroptosis was responsible for TP-induced hepatic injury. During the process of TP-induced liver damage, the Nrf2 signaling pathway was significantly suppressed. Notably, the deletion of Nrf2 in mice aggravated the extent of liver injury and ferroptosis associated with TP, whereas enhancing Nrf2 expression in cells significantly reduced TP-induced ferroptosis. Additionally, dysregulation of lipid metabolism was associated with TP-induced liver injury. TP may directly bind to Nrf2 and enhance its degradation through the ubiquitin-proteasome pathway, thereby inhibiting or reducing Nrf2 expression.
In summary, the suppression of Nrf2 by TP facilitated the occurrence of ferroptosis, resulting in liver damage.
雷公藤红素(TP)是雷公藤的主要活性成分,具有多种药理作用。然而,其潜在的肝毒性一直是临床应用中的一个重大关注点。
本研究旨在探讨铁死亡在 TP 介导的肝损伤中的作用及其机制。
本研究涉及体外和体内实验。采用 MTT 检测、肝酶测定和 H&E 染色技术评估 TP 引起的肝细胞损伤。通过测定铁水平、脂质过氧化物、谷胱甘肽(GSH)、线粒体形态以及铁死亡相关的关键蛋白/mRNA 表达来评估铁死亡。为了验证铁死亡在 TP 诱导的肝损伤中的作用,使用铁死亡抑制剂 Ferrostatin-1(Fer-1)和过表达谷胱甘肽过氧化物酶 4(GPX4)的质粒。随后,利用核因子红细胞 2 相关因子 2(Nrf2)敲除小鼠和 Nrf2 过表达质粒来探讨其潜在机制。采用非靶向脂质组学分析小鼠肝内脂质代谢。此外,还应用细胞热转移分析(CETSA)、环己酰亚胺(CHX)和 MG132 处理以及免疫沉淀(IP)实验来验证 TP 与 Nrf2 的结合及其相互作用。
TP 诱导肝细胞发生铁死亡,表现为铁积累和脂质过氧化。铁死亡是 TP 诱导肝损伤的原因。在 TP 诱导肝损伤过程中,Nrf2 信号通路受到显著抑制。值得注意的是,Nrf2 敲除小鼠肝损伤和铁死亡程度加重,而细胞中 Nrf2 表达增强可显著减少 TP 诱导的铁死亡。此外,脂质代谢失调与 TP 诱导的肝损伤有关。TP 可能通过泛素-蛋白酶体途径直接与 Nrf2 结合并促进其降解,从而抑制或减少 Nrf2 的表达。
总之,TP 抑制 Nrf2 促进铁死亡的发生,导致肝损伤。