Department of Biological Sciences, University of Alberta, Edmonton, Canada.
European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI); Hinxton, United Kingdom.
PLoS Biol. 2024 Nov 7;22(11):e3002862. doi: 10.1371/journal.pbio.3002862. eCollection 2024 Nov.
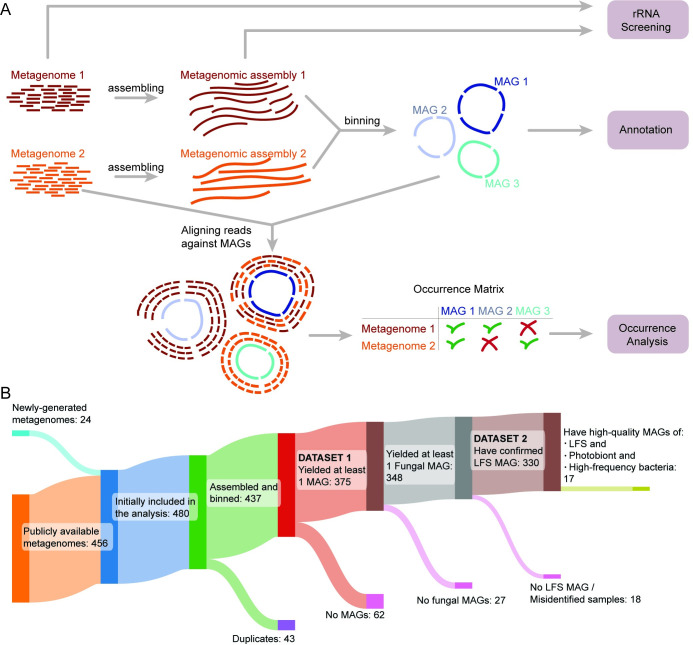
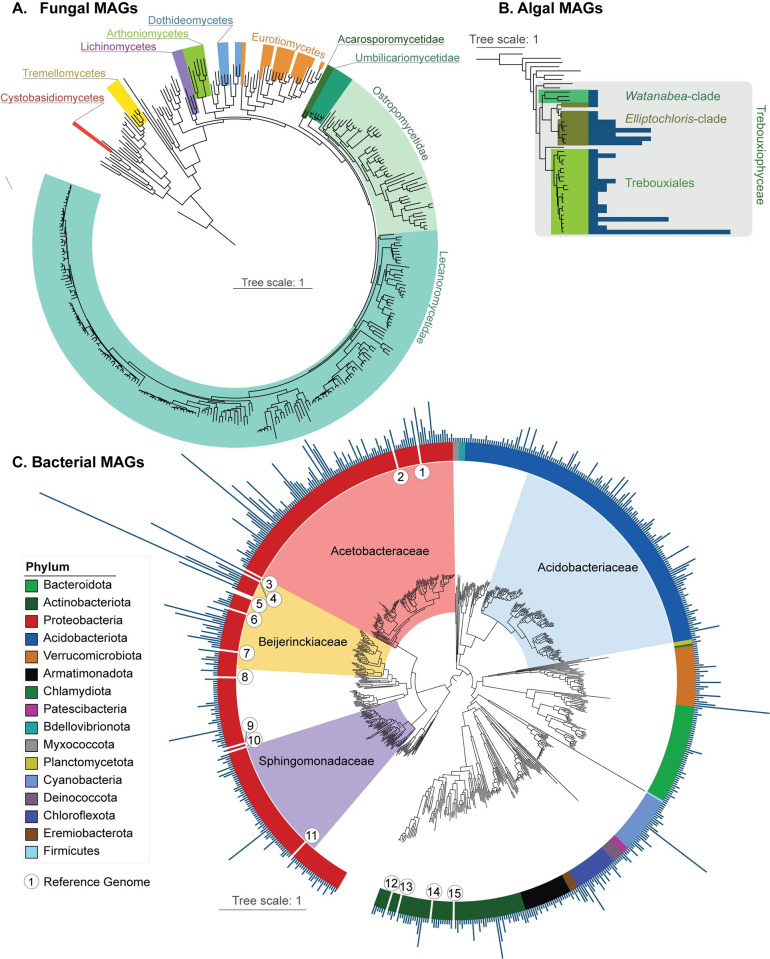
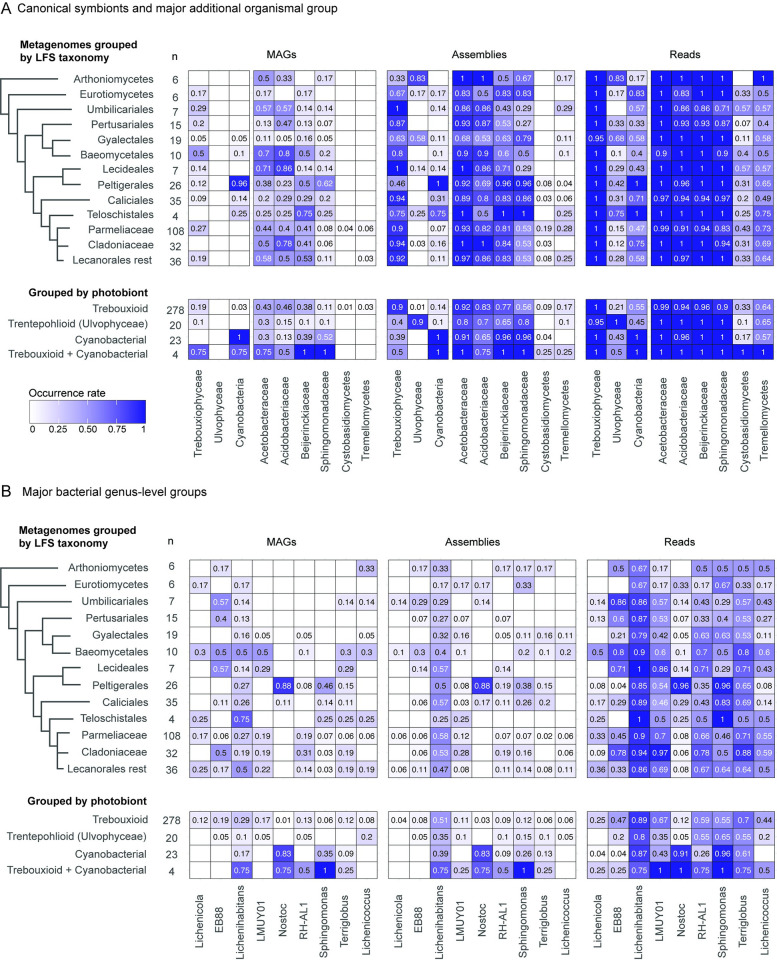
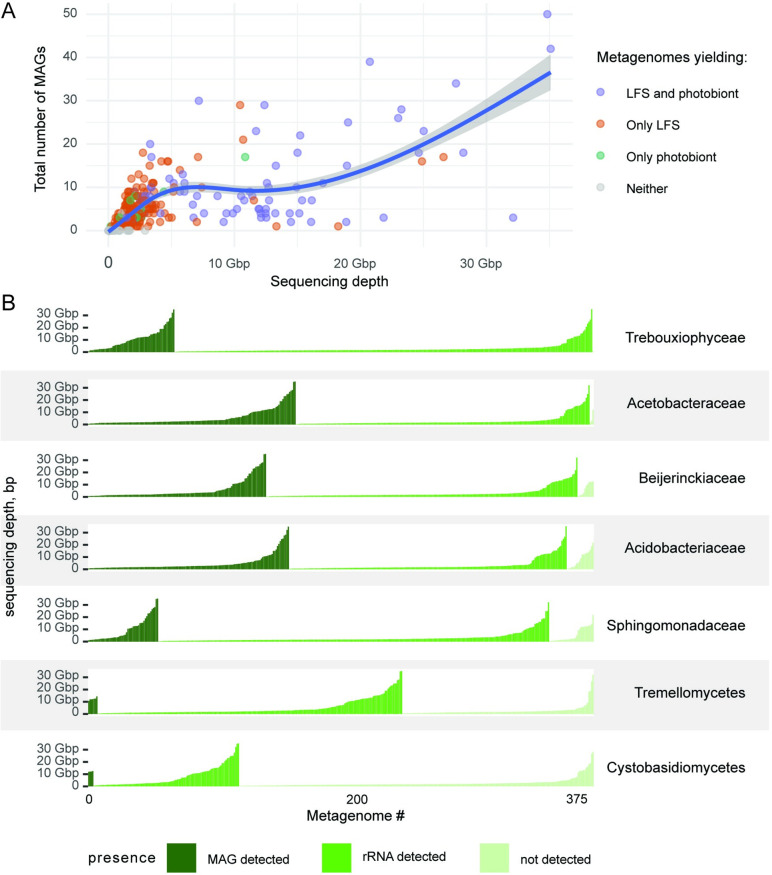
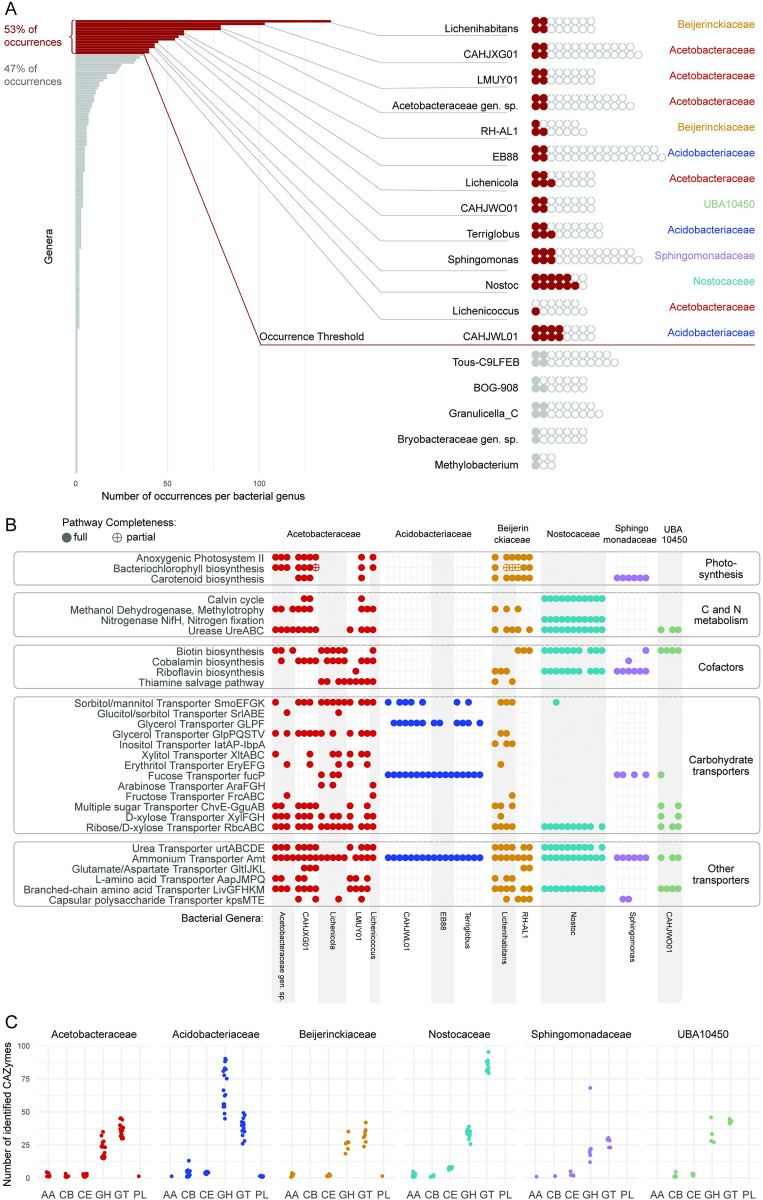
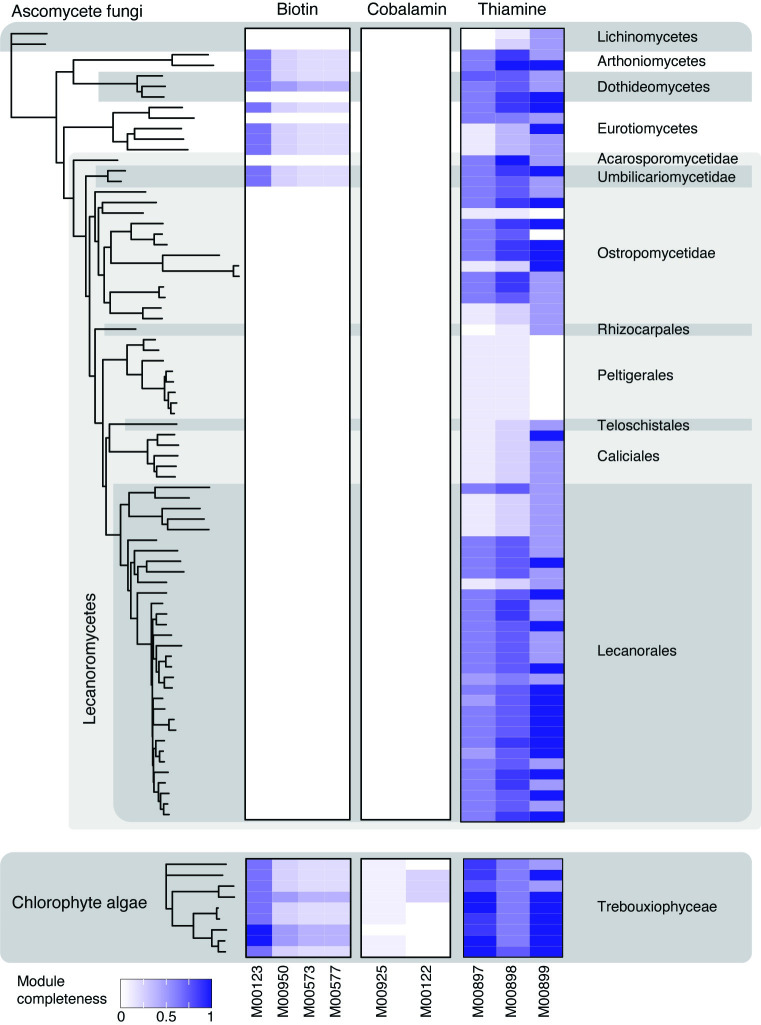
In lichen research, metagenomes are increasingly being used for evaluating symbiont composition and metabolic potential, but the overall content and limitations of these metagenomes have not been assessed. We reassembled over 400 publicly available metagenomes, generated metagenome-assembled genomes (MAGs), constructed phylogenomic trees, and mapped MAG occurrence and frequency across the data set. Ninety-seven percent of the 1,000 recovered MAGs were bacterial or the fungal symbiont that provides most cellular mass. Our mapping of recovered MAGs provides the most detailed survey to date of bacteria in lichens and shows that 4 family-level lineages from 2 phyla accounted for as many bacterial occurrences in lichens as all other 71 families from 16 phyla combined. Annotation of highly complete bacterial, fungal, and algal MAGs reveals functional profiles that suggest interdigitated vitamin prototrophies and auxotrophies, with most lichen fungi auxotrophic for biotin, most bacteria auxotrophic for thiamine and the few annotated algae with partial or complete pathways for both, suggesting a novel dimension of microbial cross-feeding in lichen symbioses. Contrary to longstanding hypotheses, we found no annotations consistent with nitrogen fixation in bacteria other than known cyanobacterial symbionts. Core lichen symbionts such as algae were recovered as MAGs in only a fraction of the lichen symbioses in which they are known to occur. However, the presence of these and other microbes could be detected at high frequency using small subunit rRNA analysis, including in many lichens in which they are not otherwise recognized to occur. The rate of MAG recovery correlates with sequencing depth, but is almost certainly influenced by biological attributes of organisms that affect the likelihood of DNA extraction, sequencing and successful assembly, including cellular abundance, ploidy and strain co-occurrence. Our results suggest that, though metagenomes are a powerful tool for surveying microbial occurrence, they are of limited use in assessing absence, and their interpretation should be guided by an awareness of the interacting effects of microbial community complexity and sequencing depth.
在地衣研究中,宏基因组越来越多地被用于评估共生体的组成和代谢潜力,但这些宏基因组的总体内容和局限性尚未得到评估。我们重新组装了 400 多个公开可用的宏基因组,生成了宏基因组组装基因组(MAG),构建了系统发育树,并在整个数据集上绘制了 MAG 的出现和频率。在回收的 1000 个 MAG 中,有 97%是细菌或提供大部分细胞质量的真菌共生体。我们对回收的 MAG 的绘制提供了迄今为止对地衣中细菌最详细的调查,表明来自 2 个门的 4 个科级谱系在地衣中的细菌出现频率与来自 16 个门的 71 个其他科级谱系一样多。对高度完整的细菌、真菌和藻类 MAG 的注释揭示了功能特征,表明维生素原营养体和营养缺陷型相互交织,大多数地衣真菌对生物素营养缺陷型,大多数细菌对硫胺素营养缺陷型,少数注释的藻类既有部分途径也有完整途径,这表明地衣共生体中的微生物交叉喂养具有新的维度。与长期存在的假说相反,除了已知的蓝藻共生体外,我们没有发现其他细菌固氮的注释。藻类等核心地衣共生体仅在地衣共生体中以已知存在的一小部分地衣共生体中被回收为 MAG。然而,使用小亚基 rRNA 分析可以检测到这些和其他微生物的存在,包括在许多原本未被认为存在的地衣中。MAG 回收率与测序深度相关,但几乎可以肯定受到影响 DNA 提取、测序和成功组装的生物属性的影响,包括细胞丰度、倍性和菌株共存。我们的研究结果表明,尽管宏基因组是调查微生物存在的有力工具,但它们在评估不存在方面的作用有限,并且应该根据对微生物群落复杂性和测序深度相互作用的认识来指导其解释。